

 分享
分享

摘要
目的:提高对先天性角化不良(DC)伴肺纤维化(PF)的认识。
方法:回顾性分析南京大学医学院附属鼓楼医院呼吸与危重症医学科2023年11月收治的1例先天性角化不良伴肺纤维化患者的临床资料及诊疗过程,并进行相关文献复习。分别以“先天性角化不良”“肺纤维化”及“先天性角化不良”“间质性肺疾病”为关键词检索中国知网、万方数据库,分别以“dyskeratosis congenita”“pulmonary fibrosis”及“dyskeratosis congenita”“interstitial lung disease”为关键词检索PubMed数据库,检索范围为2024年10月1日之前的所有中文和英文文献,纳入其中有比较详细的临床资料,包括病史、体格检查、辅助检查及诊疗经过的文献,共21篇,结合本文报道的1例患者,共收集24例患者信息。
结果:患者男,25岁,因“咳嗽伴气喘3个月余,加重1周”入院,临床主要表现为皮肤色素沉着、指(趾)甲营养不良、三系血细胞减少、间质性肺炎,基因检测Sanger测序结果显示,患者TINF2基因存在杂合突变(c.844C>T),结合临床表现及基因检测,诊断为DC。文献检索纳入的24例患者中,男20例,女4例,年龄9~52岁,PF诊断中位年龄35岁。14例(58.3%)同时有典型皮肤网状色素沉着、指(趾)甲营养不良、黏膜增生性白斑三联征的表现,7例(29.2%)仅有其中一种或两种表现,3例(12.5%)无典型的三联征表现。1例未提及血细胞情况,15例(65.2%)为全血细胞减少,5例(21.7%)为一系或两系血细胞减少,3例(13.1%)血细胞无异常。8例患者行外周血端粒长度测量,均较同年龄组人群缩短。14例患者接受端粒维持相关基因的检测,均发现有不同位点的基因突变。
结论:尽管先天性角化不良是罕见病,但依靠简单视诊就可以得到提示。当合并有典型皮肤改变、甲发育不良、骨髓衰竭的患者,尤其是年轻患者,因间质性肺炎至呼吸科就诊时,呼吸专科医生需高度怀疑本病,应尽早完善相关基因检测以明确诊断。
先天性角化不良(dyskeratosis congenita,DC)是一种罕见的由端粒调控相关基因突变所致的遗传性疾病,由Zinsser于1906年首次报道,发病率约为一百万分之一,男女发病比例13∶1,多在5~12岁时发病,无种族差异[1]。由于DC属罕见病,临床对其缺乏了解,DC肺受累更少见报道,临床资料和经验积累有限,为提高呼吸专科医生对该病的认识,减少漏诊、误诊。现回顾分析本院收治的1例DC合并肺纤维化(pulmonary fibrosis,PF)患者的临床资料及诊疗过程,并结合相关文献复习探讨。
临床资料
患者男,25岁,因“咳嗽伴气喘3个月余,加重1周”于2023年11月24日入住我院。患者于3月余前无明显诱因出现咳嗽,咳少量白色黏痰,活动后气喘明显,无发热、胸痛、咯血等不适。2023年9月至当地医院行胸部CT检查,发现双肺多发斑片状影,胸膜下为主,给予抗感染治疗(具体用药及剂量不详),症状未缓解。后至南京某三甲医院住院诊治,完善支气管镜检查,支气管肺泡灌洗液(BALF)半乳甘露聚糖抗原检测(GM试验)1.642 μg/L、支气管肺泡灌洗液新冠病毒核酸阳性,予以先诺特韦片/利托那韦片抗新冠病毒、伏立康唑抗真菌治疗,患者症状仍无明显缓解。1周前患者无明显诱因气喘加重,稍动即喘,时有鼻出血,至我院急诊就诊,查血常规示白细胞2.4×109/L,血红蛋白86 g/L,血小板28×109/L。胸部CT示双肺间质性肺炎、左侧少量液气胸、左侧锁骨上窝及纵隔少量积气、双侧肺气肿伴肺大泡、心包少量积液、两侧胸膜增厚。予以输注血小板支持治疗。为进一步明确病情,急诊以“间质性肺炎”收入院。
患者平素身体健康状况一般。既往诊断“再生障碍性贫血”10年余,近3年服用环孢素、叶酸片治疗,1个月前在南京某三甲医院住院时停用环孢素。半年前曾有双下肢摔伤史。否认吸烟饮酒史、接触化学物质及放射线史。父母体健,非近亲婚配,否认遗传病家族史。入院体检:体温36.5 ℃,脉率87次/min,呼吸频率20次/min,血压113/67 mmHg(1 mmHg=0.133 kPa),体重指数 16.67 kg/m2,意识清楚,精神状态一般,容貌消瘦,浅表淋巴结未及肿大。口腔黏膜未发现明显白斑。双肺呼吸音粗,双下肺可闻及少许捻发音。心脏、腹部体检未见明显异常。双下肢无水肿。颈部、躯干、四肢皮肤色素沉着。双下肢皮肤局部破溃、结痂,未完全愈合。双手手指、脚趾指甲萎缩(图1, 2, 3, 4, 5, 6, 7, 8)。入院初步诊断:间质性肺炎、再生障碍性贫血。
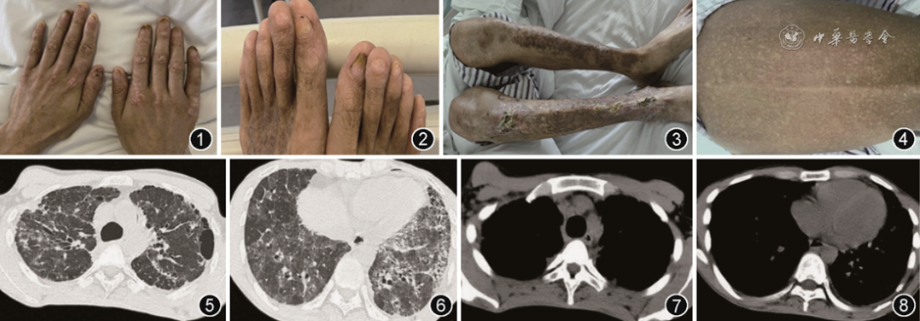
图1,2患者手指和脚趾可见甲萎缩和营养不良
图3患者双下肢皮肤可见局部破溃、结痂
图4患者背部可见网格状色素沉着
图5~8胸部CT显示患者双肺多发斑片状高密度影,边缘模糊,小叶间隔增厚,部分病灶呈网格样改变,伴局限性肺气肿,左肺上叶肺大疱纵隔少量积气,双侧胸膜轻度增厚,心包少量积液
入院后实验室检查,血常规示白细胞3.5×109/L,血红蛋白74 g/L、血小板47×109/L。红细胞沉降率95 mm/1 h。肝肾功能及心肌酶未见明显异常。凝血功能:D-二聚体0.66 mg/L,余无异常;补体+免疫常规示:IgE 1 864 IU/ml,余无明显异常;血淋巴细胞亚群计数:CD4+T细胞计数 265个/μl,余无明显异常。尿、便常规,降钙素原、EB病毒DNA、巨细胞病毒DNA、心肌酶、甲状腺功能、自身抗体、抗磷脂抗体均无明显异常。痰培养、痰真菌培养、痰找抗酸杆菌,血隐球菌荚膜抗原、β-(1,3)-D葡聚糖检测(G试验)、半乳甘露聚糖抗原检测(GM试验)及新型冠状病毒核酸检测均阴性。甲型流感病毒抗原检测阳性。动脉血气分析示(吸氧2 L/min):pH值为7.41,PaCO2 37.8 mmHg,PaO2 110 mmHg,SaO2 98%,氧合指数379。给予吸氧治疗,头孢哌酮舒巴坦抗细菌、伏立康唑抗真菌、奥司他韦抗流感病毒。患者贫血,血小板明显下降,输注红细胞悬液、血小板。
为进一步明确肺部病变性质,2024年11月30日行支气管镜检查,镜下未见特异性表现。肺泡灌洗液细菌培养、真菌培养、曲霉菌抗原、结核分枝杆菌及利福平耐药测定均阴性;涂片找抗酸杆菌阴性,涂片找弱抗酸杆菌阴性;灌洗液细胞分类示中性粒细胞30%、淋巴细胞5%、组织细胞65%,灌洗液未查见恶性肿瘤细胞。灌洗液病原学宏基因组检测示细环病毒、甲型流感病毒、人类冠状病毒229E。因患者血小板减少、气喘明显,未能行肺活检。进一步行骨髓穿刺+活检术,骨髓穿刺涂片示骨髓增生重度减低,粒红二系均增生减低,巨核细胞未见,中性分叶核细胞比例明显增高。骨髓活检示造血组织少见,造血组织增生减低,粒系红系均增生减低,巨核细胞未见。骨髓流式、染色体检查均未见异常。
患者青年男性,有全身皮肤异色病样改变,双手指甲、双足趾甲萎缩,有再生障碍性贫血病史、骨髓衰竭,间质性肺炎表现,无结缔组织病依据,考虑系统性疾病导致,先天性疾病可能性大。联系到再生障碍性贫血的可能病因为先天性角化不良[2],该疾病主要表现为皮肤色素异常、口腔白斑、指甲营养不良三联征,结合本例患者临床特征表现,考虑先天性角化不良可能性大。
取得患者及家属同意后,通过芯片捕获高通量测序进行患者及其父母、妹妹静脉血全外显子组测序,并对致病可能性较高基因变异进行Sanger测序验证。结果显示,患者TINF2基因存在杂合突变,在844号核苷酸由胞嘧啶C变成胸腺嘧啶T(c.844C>T),导致282号氨基酸由精氨酸(arginine)变成半胱氨酸(cysteine,p.R282C),该变异为先天性角化不良已知的致病突变[3, 4]。双亲及妹妹均为野生型,考虑患者致病基因为新发突变。通过定量PCR方法分析患者骨髓细胞端粒长度,为健康对照(外周血细胞)的0.792(P=0.03)。由于目前DC尚无确切有效的治疗方法,多为对症处理改善症状,患者出院后在家中吸氧,间断至当地医院输注红细胞、血小板支持治疗,体力状态较差,半年后因呼吸衰竭去世。
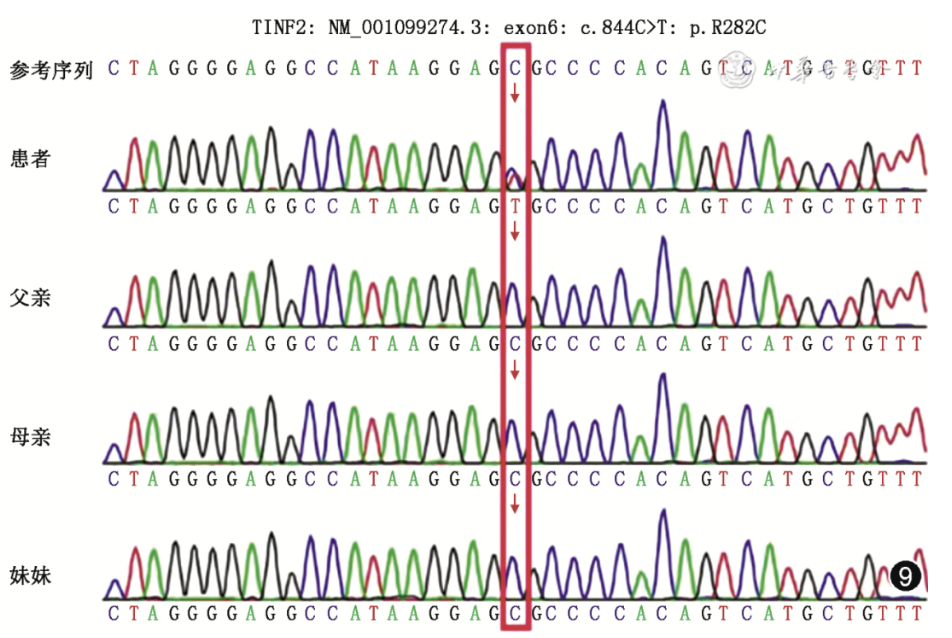
图9 患者TINF2基因上的第844位碱基发生C➝T的杂合突变,双亲及妹妹均为野生型
文献复习
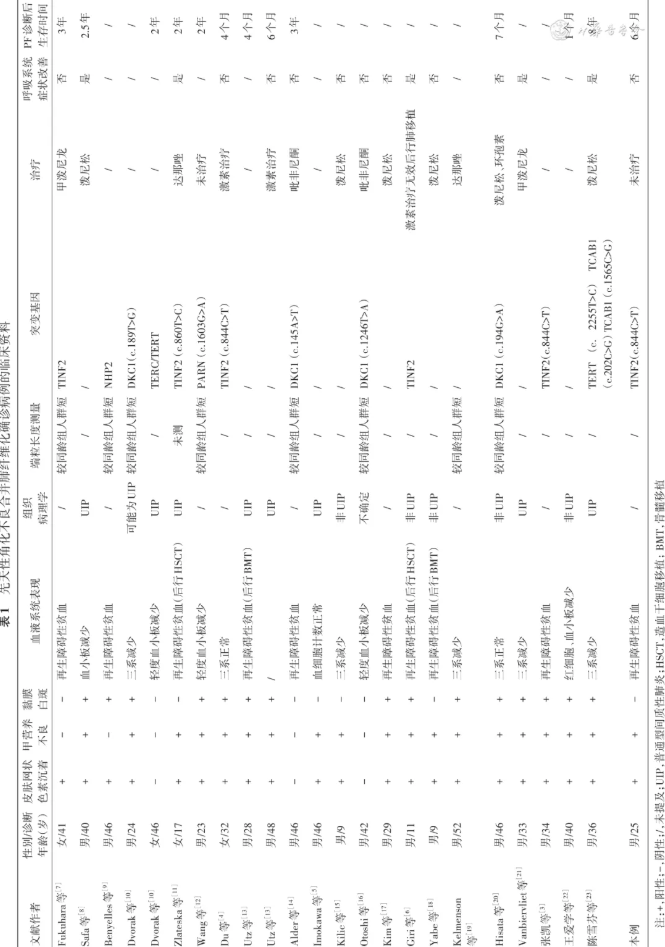
分别以“先天性角化不良”“肺纤维化”及“先天性角化不良”“间质性肺疾病”为关键词检索中国知网、万方数据库,以“dyskeratosis congenita”“pulmonary fibrosis”及“dyskeratosis congenita”“interstitial lung disease”为关键词检索PubMed数据库,纳入2024年10月1日之前发表的所有中文和英文文献,排除重复文献及部分临床资料不全文献,共获取文献21篇,均为病例报道或病例系列报道,收集病例23例。结合本文报道的1例,24例患者中男20例,女4例,年龄9~52岁,PF诊断中位年龄35岁,详见表1。24例中14例(58.3%,14/24)同时存在典型皮肤网状色素沉着、指(趾)甲营养不良、黏膜增生性白斑三联征的表现,7例(29.2%)仅有其中一种或两种表现,3例(12.5%,3/24)无典型的三联征表现。1例未提及血细胞情况,15例(65.2%,15/24)为全血细胞减少,5例(21.7%,5/24)为一系或两系血细胞减少,3例血细胞无明显异常。8例患者行外周血端粒长度测量,均较同年龄组人群缩短。14例患者接受端粒维持相关基因的检测,均发现有不同位点的基因突变,文献复习中发现,DC合并肺纤维化患者突变基因包括DKC1、TINF2、PARN、NHP2、TERC、TERT、TCAB1。1994年Imokawa等[5]首次报道DC肺受累的病理类型为普通型间质性肺炎(usual interstitial pneumonia,UIP)。我们发现在文献报道的15例基于外科肺活检或尸检的组织病理学描述中,8例(8/15)为UIP,1例有肺纤维化表现但无法明确具体病理类型,1例可能为UIP,5例为非UIP。在提供完整肺部影像学信息的21例患者中,10例可见牵拉性支气管扩张,9例可见蜂窝影,6例可见磨玻璃影,具有典型UIP影像学征象的仅13例。有详细治疗资料的共15例,单纯接受激素治疗的共8例(2例有效,6例无效),1例予激素联合免疫抑制剂治疗(无效),2例行吡非尼酮治疗(均无效),2例接受达那唑治疗(1例未提及呼吸系统症状改善情况,1例使用后肺功能有改善但2年后死于败血症),1例激素治疗无效后行肺移植[6],肺移植后随访21个月患者一般情况良好,呼吸系统症状明显改善。有精确死亡时间记录的共13例,其中PF诊断后最长生存时间为8年,最短1个月。
讨论
DC是一种端粒长度维持系统紊乱所致的先天性遗传性疾病,以X连锁隐性遗传方式为主,故患者以男性居多,少数为常染色体显性遗传或常染色体隐性遗传[1]。端粒位于真核生物染色体末端,如帽子一般,对维持染色体的稳定性和完整性有重要作用。人端粒长度为 5~15 kb,由6个碱基重复序列(TTAGGG)和由多种蛋白质组成的shelterin复合体共同构成。每次细胞分裂都会导致端粒重复序列丢失 50~200 bp,当端粒长度到达临界阈值,细胞发生凋亡或进入衰老[24]。端粒酶对端粒长度的维持起着决定性作用,在细胞分裂过程中,端粒酶通过其逆转录酶活性复制和延长端粒DNA并填补端粒复制过程中引物分离留下的空隙。研究表明,端粒或端粒酶组分的多个基因突变均可引起端粒酶含量或活性的改变,进而引起DNA复制异常,导致端粒异常缩短,促使细胞过早死亡、衰老或基因组不稳定,增加DC的患病概率[25]。目前共发现20个与DC有关的基因突变[26],包括DKC1、TERC、TERT、NOP10、NHP2、TINF2、TCAB1、RPA1、PARN、CTC1、ACD、USB1、RTEL1、NAF1、ZCCHC8、NPM1、POLA1、POT1、MDM4、DCLRE1B,然而上述基因突变仅涵盖约70%的DC患者,在约30%的DC患者身上未能发现这些基因的突变。相信随着对更多DC个体及其家系的研究,未来会有新的致病基因被发现。本例患者基因测序发现TINF2基因突变,TINF2基因编码的TIN2是shelterin复合体的一个组分,该复合体由6个亚基(TRF1、TRF2、TPP1、POT1、TIN2、RAP1)组成,故TINF2基因不直接参与端粒酶的功能,而是通过影响端粒蛋白复合体亚基导致端粒缩短。
DC可累及多个器官系统,如皮肤、黏膜、大脑、牙齿、眼睛、肺、骨髓、胃肠道、肝脏、肾脏等,并具有肿瘤易感性[27]。DC患者临床上常见典型皮肤黏膜三联征:包括皮肤网状色素沉着、指(趾)甲营养不良、黏膜增生性白斑,常在儿童期即出现,约46%的DC患者同时存在经典三联征,80%~90%的DC患者至少存在其中一种 [28],是临床早期发现该病的重要线索。DC患者常出现骨髓衰竭,造血障碍通常发生在20岁之前,早期可仅表现为一系或两系血细胞减少,随着病情进展,80%的患者在30岁前出现骨髓衰竭[29]。部分患者骨髓衰竭可出现在皮肤黏膜异常病变之前,导致初诊为“特发性再生障碍性贫血”。DC可罹患各类恶性实体肿瘤和血液系统肿瘤,有研究显示DC患癌症的风险较普通人高11倍[30]。据统计,DC患者平均存活年龄为30岁,骨髓衰竭(60%~70%)、肺部病变(10%~15%)和恶性肿瘤(10%)是DC患者死亡的主要原因[29]。
目前关于DC肺受累的患病率及发病率尚无确切统计资料。鉴于许多DC患者在早年就死亡,肺部症状尚未出现,因此DC肺部受累可能被低估。此外,部分DC患者因肺部症状就诊,但因皮肤表现轻微或临床医师对该病没有充分认识,亦会导致DC漏诊。文献报道的DC肺部表现有肺纤维化、支气管哮喘、气胸、支气管扩张、支气管肺发育不良、肺动静脉畸形、肺部感染等。DC也被认为是一种免疫缺陷综合征[31],DC患者可出现T细胞免疫缺陷、NK细胞活性下降等,是机会性感染的重要原因。肺纤维化(pulmonary fibrosis,PF)是DC的一种进展性和致命性的并发症,我们通过复习国内外文献,汇总了DC合并PF的30余例患者,年龄最小为9岁[15],最大为52岁[19],PF诊断中位年龄35岁,考虑DC肺纤维化的形成可能是一个缓慢进展的过程,故诊断年龄常较滞后。在少数DC患者中,肺纤维化可能是疾病的首发表现[14]。目前认为DC主要是由于端粒维持缺陷,导致细胞复制过程中染色体缩短和基因丢失,最终引起细胞凋亡致病,特别是在皮肤和血液这种细胞增殖较快的器官系统中。DC肺纤维化发病机制尚不清楚,最近的研究表明,端粒功能失调或端粒酶活性下降可能会影响肺泡上皮细胞损伤后的再生和修复[32]。细胞凋亡失调引起的异常修复过程在肺纤维化的发病机制中起重要作用[33]。本例患者自幼年时出现颈部、躯干、四肢皮肤色素沉着,10余岁时诊断“再生障碍性贫血”,25岁因咳嗽气喘查胸部CT提示肺间质纤维化,推测患者幼年时已经发病,发现肺部受累时已是肺纤维化表现,提示DC肺部受累发生较为隐匿。尽管DC是一种遗传性疾病,但本例患者无阳性家族史,考虑自身胚系突变产生的致病基因。本研究中4例患者于儿童时期因严重再生障碍性贫血行造血干细胞移植(hematopoietic stem cell transplatation,HSCT)或骨髓移植(bone marrow transplantation,BMT),后续随访过程仍有出现肺纤维化,考虑HSCT或BMT可以治愈DC患者的骨髓衰竭,但不能消除肺纤维化发生的风险。本例患者虽有皮肤黏膜异常表现,幼年时开始出现骨髓衰竭,但在多次就医过程中一直未被诊断出先天性角化不良,后因呼吸系统症状加重,被我院呼吸科医生识别,提示呼吸科医生应具备对可累及呼吸系统罕见病的高度警觉性。
目前国内外尚未明确DC的诊断标准,通过复习文献,该病的临床诊断主要基于典型三联征加上骨髓衰竭以及多系统受累征象。确诊检测手段包括端粒长度测量和基因检测。综合分析文献报道中DC 的主要诊断标准如下:(1)存在 4 个主要特征(皮肤网状色素沉着、甲营养不良、黏膜增生性白斑和骨髓衰竭)中的至少 2 个;(2)端粒长度较同年龄组人群显著缩短;(3)检测到与端粒或端粒酶有关的特定基因突变;(4)家族中有类似症状,遗传学分析符合X连锁隐性遗传或常染色体显性遗传或常染色体隐性遗传;大多数研究者认为符合以上两点即可确诊。本研究中24例患者均有皮肤黏膜三联征或血液系统异常表现,基因检测或端粒长度测量均有阳性发现。
DC尚无确切治疗手段,激素治疗对部分患者可能有效,有研究表明雄激素可调节端粒酶基因的表达,延缓纤维化进展[34],但缺乏大样本的临床研究,疗效有待进一步验证。对于发生肺纤维化但无多器官衰竭的DC患者可考虑肺移植。随着科学技术的不断进步,以基因疗法为代表的新一代精准医疗快速兴起,其中基因编辑技术在治疗罕见遗传病方面展现了巨大的潜力,但目前仍面临许多技术壁垒及伦理争议。
综上所述,DC是一种多系统、进行性疾病,尽管为罕见病,但具有较为明显和直观的特征性表现,依靠简单视诊就可以得到提示。当合并有典型皮肤改变、甲发育不良、骨髓衰竭的患者,尤其是年轻患者,因间质性肺炎至呼吸科就诊时,呼吸专科医生需高度怀疑本病,应尽早完善相关基因检测以明确诊断。同时,呼吸科医生应提高对肺纤维化继发因素的识别,扩展对先天性角化不良等罕见病因的认识。
参考文献(略)
作者:彭雅丽 钱夏婷 田亚琼 桂贤华 高玉娟 曹孟淑 肖永龙 李燕;第一作者单位:南京大学医学院附属鼓楼医院呼吸与危重症医学科;通信作者:李燕,南京大学医学院附属鼓楼医院呼吸与危重症医学科
引用本文: 彭雅丽, 钱夏婷, 田亚琼, 等. 角化不良合并骨髓增殖障碍三系减低[J]. 中华结核和呼吸杂志, 2025, 48(6): 540-547. DOI: 10.3760/cma.j.cn112147-20241010-00593.
本文转载自订阅号「中华结核和呼吸杂志」
原链接戳:【论著】角化不良合并骨髓增殖障碍三系减低
* 文章仅供医疗卫生相关从业者阅读参考
本文完
责编:Jerry