

 分享
分享
摘要
肺纤维化是多种异质性间质性肺疾病的终末期表现,超过200 种因素促成了这种疾病状态。近年来,特别是在2019年SARS-CoV-2爆发后,病毒感染与肺纤维化之间的关系越来越受到关注。然而,病毒诱发的肺纤维化的机制尚不完全了解。本文回顾了肺纤维化与几种病毒之间的关系,例如人类嗜T淋巴细胞病毒(HTLV)、人类免疫缺陷病毒(HIV)、巨细胞病毒(CMV)、EB病毒(EBV)、小鼠γ-疱疹病毒68(MHV-68)、流感病毒、禽流感病毒、中东呼吸综合征(MERS)-CoV,严重急性呼吸综合征(SARS)-CoV和SARS-CoV-2,并探讨了病毒感染诱发的肺纤维化的机制。这将为探索针对SARS-CoV-2等病毒引起的肺纤维化的潜在的抗纤维化治疗靶点,提供新的线索。
译者:章凯毅、李超;审核:胥武剑
关键词:病毒感染, 肺纤维化, 机制, SARS-CoV-2, 潜在的抗纤维化治疗
引言
肺纤维化是多种类型的严重肺损伤的结果,其主要与炎症反应有关。肺泡炎症反应对于增强肺部宿主防御很重要,肺泡巨噬细胞有助于这种反应[1]。病毒可能通过以下两种途径诱发肺纤维化:① 病毒感染对肺部造成直接损害。在大多数病毒感染期间,病毒会对肺部造成迅速和直接的损害。此时,伤口愈合反应被激活,然而,病毒诱导持续但异常的肺损伤和/或伤口愈合,导致肺纤维化的形成。②病毒感染引起免疫介导的损伤。肺损伤后,肺部处于炎症浸润状态,病毒感染激活了免疫系统。巨噬细胞、中性粒细胞、嗜酸性粒细胞和 Th2 细胞聚集在损伤部位并释放大量促炎和促纤维化细胞因子/因素,例如转化生长因子-β (TGF-β)、肿瘤坏死因子-α (TNF-α)、基质金属蛋白酶 (MMPs)、金属蛋白酶组织抑制剂 (TIMPs)、白细胞介素 IL-1、IL-4、IL-5、IL-6、IL-13 和 IL-17。病毒与这些因素的综合作用,诱发了持续和实质性的肺损伤,促进了肺纤维化(图1)。病毒感染诱发肺纤维化患者的预后各不相同。有些人最终死亡,而有些人则预后良好。在出院的重症COVID-19肺炎患者中,超过1/3发生肺纤维化[2]。此外,已经有许多研究报告称:巨细胞病毒、流感病毒、禽流感病毒、SARS-CoV、中东呼吸综合征冠状病毒等病毒会对肺部造成长期损害,并且在感染后很长一段时间内仍然是肺纤维化的危险因素。本文中,我们回顾了多种病毒感染与肺纤维化的关系,并总结了潜在机制以及未来的研究方向。这些机制可能为包括SARS- CoV-2在内的病毒诱发的肺纤维化的治疗策略提供新的思路。
人类嗜T淋巴细胞病毒
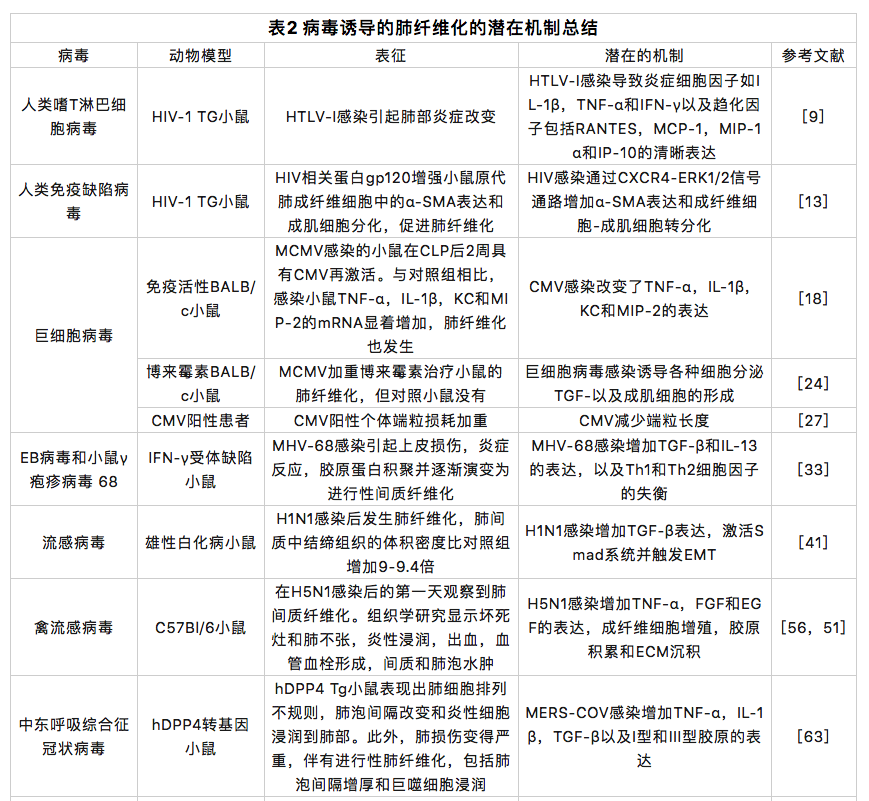
HTLV-I型是首个报道的人类致癌逆转录病毒,是成人T细胞白血病淋巴瘤(ATL)和HTLV-I相关脊髓病(HAM)/热带痉挛性截瘫(TSP)的病原体[3]。肺部是受HTLV-1介导的炎症影响的器官之一,炎症涉及间质性肺炎、细支气管炎和肺泡炎。通过对106例HTLV阳性患者的计算机断层扫描(CT)的研究发现,65例(61.3%)患者的CT有异常结果,包括阴影磨玻璃影(31.1%)、支气管扩张(26.4%)、小叶中心性结节(23.6%)、小叶间隔增厚(17.9%)、蜂窝肺(4.7%)和铺路石征(2.8%)[4]。两项CT研究表明,HTLV患者会发生肺纤维化[4,5](表1)。HTLV-I的病毒转激活蛋白Tax激活了许多细胞基因,如TGF-β,这可能是HTLV-I诱导的肺纤维化的一个重要原因[6]。Teruya 等人的研究表明,HTLV-I感染肺上皮细胞,并通过诱导NF-κB和激活蛋白(AP)-1刺激它们产生细胞因子、趋化因子和细胞粘附分子,促进了HTLV-I相关肺部疾病的临床表现[7]。Matsuyama等人证明,与HTLV-I阴性患者相比,患有隐源性纤维化肺泡炎(CFA)的HTLV-I阳性患者,支气管肺泡灌洗液(BALF)中的MMP-2水平显著增加,且TIMP-2没有差异,这表明MMP和TIMP的不平衡可能是HTLV诱导的肺纤维化的原因。他们还观察到巨噬细胞炎症蛋白(MIP)-1α、干扰素γ诱导蛋白(IP)-10和可溶性细胞间粘附分子(sICAM)在BALF中表达增加[8]。Miyazato等人表明,HTLV-I感染导致炎症细胞因子的明显表达,如IL-1β,TNF-α和干扰素(IFN)-γ以及趋化因子,包括激活时调节的正常T细胞表达和分泌(RANTES),单核细胞化学引诱蛋白1(MCP-1),MIP-1α和IP-10在mRNA水平的转基因小鼠的肺部[9](表2)。


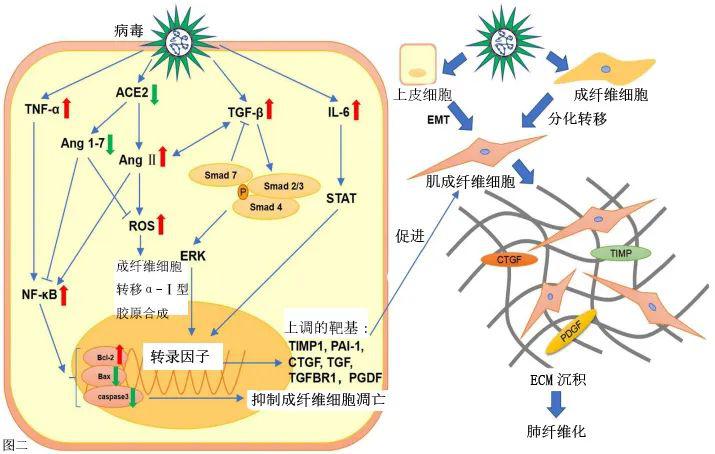
人类免疫缺陷病毒
肺部疾病是 HIV 感染的常见并发症,通常表现为间质性肺炎。HIV诱发间质性肺疾病的高分辨率计算机断层扫描(HRCT)显示阴影磨玻璃影、实变和蜂窝影,组织病理学特征为间质浸润[10](表1)。Crothers等人表明,与未感染者相比,感染HIV的患者发生肺纤维化的可能性更高[11]。此外,Leader等人发现,29.4%的HIV患者肺部至少有一定程度的纤维化样改变,这种改变与HIV病毒载量和吸烟状况呈显著正相关[12]。然而,由于缺乏随访研究,尚不清楚HIV诱导的肺纤维化是进行性的还是稳定的。在基础研究中,Marts等人发现HIV蛋白gp120通过激活小鼠原代肺成纤维细胞中的CXCR4-ERK1/2信号通路来诱导α平滑肌肌肌动蛋白(α-SMA)的表达和成纤维细胞向肌成纤维细胞的转分化,从而促进肺纤维化。同时,他们注意到HIV转基因小鼠的肺部显示羟脯氨酸呈年龄依赖性增加,羟脯氨酸是胶原蛋白合成所需的氨基酸[13](表2)。这可能提示老年HIV患者中肺纤维化形成的机制。此外,HIV刺激巨噬细胞、血小板和淋巴细胞产生内皮素-1、IL-1β、IL-6和TNF-α等细胞因子[14]。TNF-α通过NF-κB通路和上调TGF-β、血小板衍生生长因子(PDGF)-α和PDGF-β的表达来加重炎症并促进肺纤维化[15,16]。此外,NF-κB通过上调Bcl-2以及下调Bax和半胱天冬酶3来抑制成纤维细胞凋亡,以促进肺纤维化[17]。这些促炎细胞因子已被证明在肺纤维化的发展中起重要作用,其上调表达可能是HIV诱导的肺纤维化的重要因素(图2)。
巨细胞病毒
CMV是疱疹病毒家族的成员,在宿主初次感染期间持续处于潜伏状态。完整的病毒基因组驻留在宿主中,不产生完整的感染性病毒颗粒,直到一些刺激诱导重新激活[18]。CMV再激活在非免疫抑制的危重症手术患者中可引起严重后果,间质性肺炎是常见的表现[19,20]。Yonemaru等发现,在特发性肺纤维化(IPF)或间质性肺炎中,CMV Ig M阴性后CMV免疫球蛋白(Ig)G和补体结合滴度升高[21],表明CMV感染与肺纤维化的形成相关。HRCT显示巨细胞病毒肺炎表现为双肺磨玻璃样衰减、气腔实变和多发小结节 [22](表1)。CMV再激活常与纤维蛋白性肺泡炎和轻、中度纤维化有关[23]。
在动物研究中,Cook等人表明,与对照组相比,感染MCMV的小鼠在盲肠结扎和穿刺(CLP)后2周内重新激活了CMV,TNF-α、IL-1β、趋化因子KC(KC)和MIP-2的mRNA水平发生改变,还形成了肺纤维化[18](表2)。Li等人发现,单纯感染MCMV的小鼠未发生肺纤维化。相较于对照组MCMV可加重博来霉素诱导的肺纤维化。他们还观察到蛋白水平上vimentin和phospo- Smad2的增加[24](表2),这表明TGF-β / Smad途径被激活并且Smad2 / 3在此过程中被磷酸化。磷酸化的Smad2/3与Smad4形成三聚体,然后转移到细胞核并与Smad结合元素结合,以调节TGF-β靶基因表达。TGF-β/Smad途径增加了促纤维化细胞因子的表达,如TIMP-1、纤溶酶原激活物抑制剂-1(PAI-1)、结缔组织生长因子(CTGF)、TGF和TGFR1,以激活肌成纤维细胞。同时,大量胶原蛋白的分泌,导致细胞外基质(ECM)沉积和肺纤维化的发展[25]。此外,IL-1β会导致炎症,并刺激成纤维细胞合成胶原蛋白和纤维蛋白以促进肺纤维化[26]。此外,潜伏性CMV会缩短端粒长度[2](表27),而短端粒被认为是肺纤维化的危险因素[2]。
EB病毒和小鼠γ疱疹病毒 68
除巨细胞病毒外,其他疱疹病毒也与肺纤维化有关,如EBV和MHV68。Ankermann等报告,一名12个月大的免疫功能正常的女孩在感染EBV后出现非特异性间质性肺疾病和肺纤维化。14 月龄时的 HRCT 扫描显示,中叶和右非典型下叶叶段有节段性空气潴留,中叶线状非叶段性(跨肺叶)阴影阴影,表明有局灶性瘢痕形成,内侧段和上段有支气管周围实变。开胸肺活检显示左下叶有非典型局灶性间质支气管周围浸润,伴有组织细胞和淋巴细胞以及间质纤维化和胶原组织增多。在使用吸入和口服类固醇联合治疗后,患儿运动耐量正常,休息和运动后呼吸时,动脉血氧饱和度高于93%[29](表1)。在EBV感染的上皮细胞中,Malizia等人观察到TGF-β表达增加,更昔洛韦治疗能降低其表达[30]。Sides等人证明EBV在潜伏期表达潜膜蛋白(LMP)-1,这可能通过NF-κB和ERK途径促进肺纤维化[31]。同样,Krug等人发现,抑制NF-κB信号传导会抑制纤维细胞募集趋化因子MCP-1和CXCL12的表达,以改善MHV-68诱导的促纤维化事件[32]。Mora等人证明,MHV-68感染在IFN-γ受体缺陷的小鼠中会导致其上皮损伤、炎症反应、胶原蓄积并逐渐演变为进行性间质纤维化。感染后小鼠的细胞因子谱和组织病理学特征包括TGF-β和IL-13表达增加,Th1和Th2细胞因子失衡,肌成纤维细胞病灶,II型肺泡上皮细胞增生,表面活性蛋白改变和血管改变。此外,TGF-β表达水平升高与肺纤维化进展相吻合[33]。IL-13通过多种机制诱导成纤维细胞转分化为肌成纤维细胞:(1)调节JNK信号;(2)通过抑制COX表达和前列腺素E2(PGE2)的产生来促进成纤维细胞增殖;(3)通过上调AKT信号传导中YY1(阴阳1)表达促进成纤维细胞的分化[26]。这些结果表明,TGF-β和IL-13可能在疱疹病毒诱导的肺纤维化中起重要作用。此外,疱疹病毒诱导的肺纤维化在类固醇和抗病毒治疗后消退[30,34]。在病毒感染的早期进行IFN-γ治疗可能有助于降低肺纤维化的严重程度。

流感病毒
病毒性肺炎和病毒抗原的表达与伴随的弥漫性肺泡损伤(DAD)是2009年大流行性甲型流感(H1N1)病毒感染的突出特征[35]。H1N1可迅速进展为急性呼吸窘迫综合征(ARDS),并可能诱发肺纤维化。继发于2009年大流行性流感的ARDS表现为DAD和肺透明膜的形成,以及其他组织学特征,包括支气管肺泡肺炎、间质间隔增厚、II型肺泡细胞增生、纤维化和鳞状化生[36](表1)。在一项针对 56 名 H1N1 患者的 HRCT 研究中,最常见的 CT 发现是第一周阴影磨玻璃影伴或不伴实变影。这些异常在第二周达到顶峰,随后消失,导致残留病灶在 第4 周或更晚显著减少。初始CT扫描显示阴影磨玻璃影和/或实变异常往往完全消退或导致残留病灶显著减少[37]。H1N1诱导的肺纤维化似乎具有自我康复的能力,表明其潜在机制可能与其他肺部疾病不同(表1)。Mineo等人证明,肺纤维化可能呈现不同的空间分布和时间趋势[38]。在一项为期1年的随访研究中,Saha等人证明,从H1N1诱导的ARDS中康复的患者仍有部分会发生肺纤维化。他们还发现,通过吡非尼酮、阿奇霉素和泼尼松龙的联合治疗可使患者从肺纤维化中恢复[39]。H1N1患者的肺纤维化可能与炎症后修复途径中成纤维细胞活性增加有关。在此过程中,TGF-β和胶原蛋白起着重要作用。许多研究表明,肺纤维化患者的TGF-β明显升高。Wen等人发现,除TGF-β外,观察期内其他所有细胞因子几乎没有变化。此外,他们表明TGF-β在重症患者中过量产生,但在轻度患者中没有[40],这表明肺纤维化可能与疾病的严重程度有关。此外,H1N1感染的动物模型显示出类似的结果。Shatskaya 等人发现 H1N1 感染增强了巨噬细胞和肺泡细胞的 TGF-β 和 Smad-2表达,表明 H1N1 可能通过激活 TGF-β/Smad 通路诱导肺纤维化[41](表2,图2). 此外,Drakopanagiotakis等人证明,肺泡上皮细胞凋亡增加和成纤维细胞凋亡减少在肺纤维化的发病机制中起重要作用[42]。Roberson等人发现,甲型流感病毒感染可诱发内质网(ER)应激,并引发多种反应,如半胱天冬酶-12依赖性上皮凋亡和JNK1依赖性TGF-β表达[43]。这些结果表明,流感感染可能激活TGF-β/Smad通路和ER应激,促进肺纤维化。
禽流感病毒
据报道,禽流感病毒也可诱发肺纤维化。在这里,我们主要讨论H7N9和H5N1诱导的肺纤维化。通过HRCT图像发现,所有H5N1和H7N9的幸存者均有肺部累及,阴影磨玻璃影和肺叶实变是最典型的胸部CT表现,这可能是由于DAD伴蛋白性渗出物所致,偶见细胞增大大和肺泡内出血[44,45,46](表1).在H5N1感染的早期阶段,病变在几天内迅速发展,导致双肺播散性渗出变化。在高峰阶段,大部分肺部区域受累。对于重度H5N1患者,恢复是一个漫长的过程,即使在临床症状消失后,肺部病变仍然存在[44]。在同一项研究中,重症H5N1患者的第12个月和第24个月的随访CT仍显示毛玻璃样阴影,明显的网状影,不规则的线性阴影阴影,小叶间隔增厚和小叶内线。另一名患者第12个月的随访CT仅显示极少量的残余纤维条索,这表明H5N1诱导的肺纤维化是可逆的。严重H7N9感染患者的康复过程也持续很长时间。在发病后 32 周的随访检查中, CT 图像显示不同程度的继发性纤维化和牵引性支气管扩张。此外,值得注意的是,在胸部CT结果中,淋巴细胞减少的恢复时间与恢复正常的时间高度一致,表明淋巴细胞减少是H7N9的一个特征,从中恢复有助于清除病毒和改善病情[45]。在H5N1患者的尸检中显示存在机化性DAD伴间质纤维化[47]。在H7N9感染患者的尸检报告也可见类似的表现,包括II型肺泡 细胞增生和脱落、严重肺纤维化、大量淋巴细胞浸润和少量中性粒细胞浸润,与放射学发现一致[48]。在感染H5N1的小鼠身上观察到类似的人类病理生理学。Qiao等人证明,感染H5N1的小鼠在感染7天后发生弥漫性肺炎,伴有炎性细胞浸润。在第30天,在大多数小鼠身上观察到不同程度的肺纤维化[49]。研究表明,H5N1感染显著增加了小鼠体内TNF-α、IL-6、成纤维细胞生长因子(FGF)和表皮生长因子(EGF)等促纤维化因子的分泌[50,51](表2)。此外,H5N1和H7N9感染患者的血管紧张素转换酶2(ACE2)表达降低,诱导血管紧张素II(Ang II)表达水平升高[52,53]。Ang II和ACE2是肾素-血管紧张素系统(RAS)的组成部分,该系统调节肺纤维化的发生。Ang II与Ang II型1受体(AT1R)结合,增加TGF-β,NF-κB和PAI-1的表达,以促进肺纤维化。此外,Ang II显著增加了肺成纤维细胞中NOX4水平和活性氧(ROS)的产生,通过RhoA/Rock途径刺激成纤维细胞迁移和α-胶原蛋白I的合成。相反,ACE2产生Ang1-7,通过减少NF-κB的分泌和抑制NOX4衍生的ROS介导的RhoA/Rock途径来减少炎症的相关性,以保护肺部免受损伤并抑制纤维化。ACE2对Ang II也有抑制作用[17,54]。Ang II通过线粒体途径诱导肺泡上皮细胞凋亡,并通过NF-κB途径抑制成纤维细胞凋亡[55](图2)。这些研究表明,禽流感感染降低了ACE2的表达及其对Ang II的抑制作用,可能使RAS的平衡向促纤维化方向偏移。Zou等人还证明,人重组ACE2通过减少病毒复制而有效地保护了感染H5N1小鼠的肺部组织[52]。Kuba等人发现,ACE抑制剂可在博来霉素诱发的肺纤维化小鼠中抑制其上皮细胞凋亡、间质纤维化和胶原沉积[56]。
中东呼吸综合征冠状病毒
MERS-CoV属于冠状病毒属,其受体是二肽基肽酶4(DPP4)。症状出现在MERS-CoV感染后2-14天内,轻度患者会出现低烧、流鼻涕、喉咙痛和肌肉酸痛,而重症患者则出现急性呼吸窘迫综合征。从重症患者的胸部X光片和CT扫描中,观察到多叶空腔疾病、阴影磨玻璃影和胸腔积液[57](表1)。在对一名MERS-CoV感染患者的尸检中,主要的肺组织学模式为DAD,伴有毛细支气管上皮剥落、明显的透明膜、肺泡纤维蛋白沉积和II型肺泡细胞增生[58]。Das等人表明,从MERS-CoV感染中康复的患者中有很大一部分会发展为肺纤维化,并且进展相当快速,出院后肺纤维化的平均值±SD,中位数和肺纤维化进展范围分别为82.4±66天,44天和32~230天。此外,肺纤维化的发病率和程度与MERS-CoV感染的严重程度和感染期有关[59]。MERS-CoV具有比SARS-CoV更广泛的组织嗜性,并且在抑制干扰素产生方面类似于SARS-CoV[60,61]。与SARS-CoV感染相比,MERS-CoV感染诱导IL-8、IL-12、IP-10/CXCL-10和MCP-1/CCL-2的表达水平显著升高[60]。Yeung等人证明,MERS-CoV通过增加Smad7和FGF2的表达诱导细胞凋亡[62]。Kim等人表明,在MERS-CoV感染的hDPP4转基因(hDPP4-tg)小鼠中观察到肺纤维化的组织病理学特征,包括肺泡间隔增厚、炎性单核细胞浸润、巨噬细胞极化、急性肺部炎症反应,以及促炎因子和促纤维化因子表达增加,如TNF-α,IL-1β,TGF-β,I型胶原蛋白和III型胶原蛋白[63](表2).Falzarano等人证明,MERS-CoV感染上调了IL-2、IL-4、IL-6、IL-17、IL-22、IL-27受体,并下调了IL-1、IL-12、IL-18和IL-20受体[64]。他们还注意到IFN-γ及其受体在多个时间点下调,但没有IFN-β的表达。缺乏干扰素和分泌促炎细胞因子可能会加重肺损伤并促进肺纤维化。IL-4、IL-6和IL-17还通过促进胶原合成和成纤维细胞转化为肌成纤维细胞而促进肺纤维化[26]。
严重急性呼吸系统综合症冠状病毒
SARS是一种急性传染病,由冠状病毒SARS-CoV引起,具有高发病率和死亡率[65]。SARS-CoV也属于冠状病毒属。SARS-CoV感染的症状包括持续高热、发冷、不适、肌痛、头痛和干咳。大多数患者在1-2周内康复,然而,约1/3的患者在SARS-CoV感染后出现ARDS且死亡率较高[66]。在SARS发展的急性阶段(感染后7-10天),肺部表现出DAD的特征,包括广泛的水肿,透明膜形成,肺泡塌陷和肺泡上皮脱落。在SARS发展的下一阶段(感染后10-14天),出现了纤维化的特征。随着SARS-CoV感染的发展持续2-3周以上,肺纤维化的程度也进一步加重[67,68](表1)。在死亡病例的尸检中也可以发现纤维化病变,肺纤维化的程度与SARS-CoV感染的持续时间有关。该病毒在尸检的肺组织中持续存在,表明感染后肺部持续受损[68]。肺纤维化不仅发生在SARS-CoV感染期间,而且发生在恢复期间。出院一个月后,通过HRCT发现SARS-CoV IgG阳性且肺弥散异常的51例患者中有40例发生了肺纤维化[69]。此外,与未感染者相比,从SARS-CoV感染中康复的患者,特别是高龄和病程严重的患者,肺纤维化发病率更高,肺功能更差[70]。一项针对SARS患者的15年随访研究显示,71名患者中有27名出现阴影磨玻璃影或条索样实变影,这表明肺损伤持续了很长时间[71](表1)。此外,发现一些患者中发现肺纤维化自发消退,这表明SARS-CoV感染诱导的肺纤维化是可逆的,但机制尚不清楚[69,72](表1)。尽管SARS-CoV诱导的肺纤维化的确切机制仍未确定,但各种动物研究表明,TGF-β和ACE2可能发挥重要的调节作用。如上所述,TGF-β通过多种机制促进肺纤维化。ACE2已被证明具有抗纤维化作用,并通过负调节Ang II对肺部产生保护作用(图2)。Rockx等人发现,在SARS-CoV感染的小鼠中,ACE2表达显著降低,并与死亡率有关。虽然本研究未检查肺纤维化,但促纤维化细胞因子的表达显著增加[73](表2)。Zhao等人证明,N蛋白不仅通过促进Smad-p300复合物的形成来增加TGF-β的转录反应,而且还增强了PAI-1的表达并减弱Smad3/Smad4介导的细胞凋亡[74]。同样,Gralinski等人发现,SARS-CoV感染显著提高了小鼠中促炎细胞因子,如IL-1β、TNF-α、IL-6和促纤维化TGF-β、CTGF和PDGF的转录物[75](表2)。他们还注意到PAI-1的表达增加,这诱发了肺泡II型细胞衰老、胶原沉积和促纤维化介质分泌促进肺纤维化[75,76,77](图2)。Page等人发现SARS-CoV感染导致STAT1基因剔除小鼠肺部Th2型免疫偏倚选择性活化的巨噬细胞(AAM)的分化,导致受损部位巨噬细胞过度活跃,并可能促进肺纤维化[78]。此外,Th2型免疫偏倚通过促纤维化和促炎细胞因子加重肺纤维化[79]。
严重急性呼吸系统综合症冠状病毒-2
随着COVID-19的全球暴发,肺纤维化作为SARS-CoV-2感染患者的并发症之一,值得更多关注[80]。它与疾病严重程度、年龄、ARDS、住院时间较长、心动过速、无创机械通气和较高的初始胸部CT评分有关[2,82,83]。在SARS-CoV-2感染的致死病例中,尸检时通常存在肺纤维化[84]。一名无肺部疾病史的老年女性患者在治愈 COVID-19 感染后死于严重的双侧肺纤维化。CT显示两侧广泛肺纤维化,显微镜检查显示纤维化伴蜂窝状重塑和支气管化生[85]。一项为期6个月的随访研究表明,肺纤维化不仅发生在SARS-CoV-2感染过程中,而且在超过三分之一的重症COVID-19肺炎感染患者中,在出院后阶段也发生了肺纤维化[2]。另一项随访研究表明,薄层胸部CT显示,397名新冠肺炎患者的肺纤维化在发病120天后约有三分之一可以逆转[86](表1)。尽管肺纤维化将是SARS-CoV-2感染患者康复期的主要并发症,但目前关于这种后遗症患病率的数据尚不清楚,需要时间更长、样本量更大的随访研究。COVID-19患者胸部CT最常见的模式是阴影磨玻璃影和双侧斑片状阴影[81]。在随访CT中,在3-14日内观察到毛玻璃和纤维条索影增加[87]。肺活检组织的组织病理学检查显示,双侧急性改变伴DAD、反应性II型肺泡细胞和巨噬细胞增生、斑片状炎性细胞浸润和松散间质纤维化(表1)。所有放射学和组织病理学证据表明SARS-CoV-2感染可能是肺纤维化的诱因,并且可能涉及细胞因子风暴引起的过度免疫反应。此外,Hu等人发现IFN-γ的基线水平与出院时COVID-2纤维化体积的增加呈负相关[89]。Valdebenito等人注意到,巨噬细胞在自体肺部的浸润增加,肺泡细胞大量死亡,成纤维细胞增殖[90]。
持续性肺损伤被认为是肺纤维化的主要原因。在SARS-CoV-2感染期间,病毒迅速复制并与肺上皮细胞表面的ACE2受体结合。然后II型肺泡细胞被感染,导致肺泡损伤和气体交换受损。巨噬细胞在受伤后被激活和聚集,以减少炎症。然而,促炎细胞因子和趋化因子的剧烈反应、病毒的持续增殖以及大量肺细胞的破坏等不利条件导致M1型巨噬细胞极化并进一步促进炎症。随着损伤的进展,损伤部位可观察到一系列宿主炎症反应,如细胞因子生成过多和炎症细胞的大量聚集[93,94,95]。Harrison等人表明,SARS-CoV-2感染增加了在BALF和血清中IFN-I、IFN-II、TNF-α、IL-1β、IL-6、IL-8、IL-18、GM-CSF、CXCL9、CXCL10、CXCL11、CCL-2和MIP1a的表达,导致持续性肺损伤[93]。最终,严重的瘢痕形成和肺组织纤维化是由胶原蛋白沉积引起的。Chen等人发现,与中度患者相比,重症患者表达的IL-2R、IL-6、IL-10和TNF-α水平更高[96]。IL-6已被证明通过STAT途径促进肺纤维化[97](图2),因此IL-6可能是SARS-CoV-2诱导肺纤维化的重要因素之一。Xu等人发现,SARS-CoV-2诱导的肺纤维化患者的肺泡上皮细胞中,TGF-β、纤维连接蛋白1(FN1)和CTGF等促纤维化细胞因子的mRNA转录显著增加[98]。SARS-CoV-2感染还会导致肺纤维化相关蛋白的异常表达,例如发现MMP2、MMP8和组织蛋白酶上调,E-钙粘蛋白下调[99]。此外,与SARS-CoV一样,SARS-CoV-2感染会降低ACE2表达,破坏RAS的平衡,并可能促进RAS向炎症和纤维化的方向进展[100]。
在动物模型中,SARS-CoV-2感染还诱发了促纤维化细胞因子的表达和肺纤维化的发生[91,92](表2)。虽然具体机制需要进一步研究,但SARS-CoV-2诱导的肺纤维化与其他病毒诱导的肺纤维化有许多相似之处。根据现有研究,SARS-CoV-2感染患者肺纤维化的发展伴随着TGF-β,TNF-α,IL-1β,IL-6和IL-13等细胞因子表达的增加。此外,一些研究报告称,SARS-CoV-2感染不仅会降低ACE2的表达,还会抑制自噬。所有这些都表明,TGF-β、TNF-α、IL-1β、IL-6、IL-13、ACE2和自噬抑制可能是SARS-CoV-2诱导肺纤维化的关键因素。

讨论和未来展望
虽然病毒感染诱发肺纤维化的潜在机制尚未完全阐明,但存在共同点。在大多数病毒诱导的肺纤维化中可以看到TGF-β的异常表达。同时,病毒诱导的异常免疫反应在肺纤维化的形成中也起着至关重要的作用。肺部受到病毒感染的损害,通常表现为炎症浸润,而巨噬细胞在受损部位的聚集并极化,释放大量促炎和促纤维化细胞因素/因子,如IL-1β、IL-6、IL-17和TNF-α。同时,2型免疫反应被激活并导致Th2细胞因子的分泌增加如IL-4,IL-5和IL-13。这些细胞因素/因子不仅对肺组织造成持续损伤,而且还诱发肺纤维化的形成。此外,病毒感染通常会导致ACE2的表达降低,使RAS的平衡向促纤维化方向偏移。
还值得注意的是,病毒感染常伴有自噬抑制,这导致成纤维细胞的大量增殖,促进肺纤维化的形成。SARS-CoV和MERS-CoV产生的病毒膜相关木瓜蛋白酶样蛋白酶2(PLP2-TM)诱导自噬体形成,并阻断自噬体与溶酶体的融合[101]。据报道,SARS-CoV-2通过阻碍自噬体-溶酶体融合来阻止自噬进展[102]。HIV蛋白Nef通过与自噬调节因子Beclin-1相互作用来抑制自噬过程,从而保护HIV免于降解[103]。此外,CMV编码TRS1和IRS1蛋白质,以抑制自噬过程[104,105]。MHV-68 编码M11蛋白质以拮抗自噬[106]。先前的研究还表明,异常的自噬活动会导致肺纤维化的发生。Hill等人认为,自噬抑制诱导肺泡上皮细胞的EMT,并通过异常的上皮-成纤维细胞串扰促进肺纤维化[107]。此外,病毒感染增加 ROS 表达和 mTOR 的分泌来抑制自噬,并通过 PI3K-Akt-mTOR 途径促进肺成纤维细胞增殖。ER应激不仅会增加成纤维细胞的增殖和迁移,还会损害肺泡上皮细胞中的蛋白质稳态,从而促进肺纤维化[108]。所有这些都表明,自噬减少可能是病毒感染诱发肺纤维化的一种机制。从这个意义上说,自噬调节剂可能有效治疗由包括SARS-CoV-2在内的病毒诱导的肺纤维化。
值得注意的是,病毒感染后的肺纤维化可能不是感染的直接结果,呼吸机引起的损伤等其他因素也可能发挥作用。许多重症病毒感染的患者在治疗期间使用了机械通气。随访研究表明,机械通气时间较长的患者发生肺纤维化的概率更高,预后更差[2,19]。Diem等人表明,在机械应力下的肺泡细胞可以产生信号分子与邻近细胞通信并促进纤维化反应[109]。
与SARS-CoV和其他病毒类似,SARS-CoV-2也会导致长期肺损伤和持续性的肺纤维化。为了使患者预后更好,及时的抗病毒和抗纤维化治疗是必要的。传统抗纤维化药物(即皮质类固醇)的安全性、剂量和干预时间需要进一步探索。此外,IL-6中和抗体和人重组ACE2已被证明可抑制博来霉素诱发的小鼠肺纤维化。这些干预措施是否能有效治疗SARS-CoV-2诱发的肺纤维化有待进一步研究。
病毒诱发的肺纤维化机制需要深入研究。比较病毒诱导的肺纤维化与其他类型的肺纤维化,可能会揭示其独特的特征和机制。此外,系统地比较病毒感染动物与感染后不同阶段对照动物之间的肺基因组和细胞因子谱,不仅可以提供生物标志物和治疗靶点,还可以提供早期诊断和监测纤维化进展的策略。
还有许多问题有待进一步研究。例如,由某些病毒诱导的肺纤维化会自发消退,而其他一些病毒可能会持续存在并需要药物治疗。长期的随访研究是必要的。目前,缺乏对某些病毒诱发的肺纤维化的随访研究。对患者进行定期HRCT检查将有助于确定肺纤维化是否可逆,以及持续多长时间。病毒通常在感染的早期阶段迅速损害肺部。因此,抗病毒治疗联合抗纤维化治疗,可能最好在早期进行。为此,需要更明确的标准来确定病毒诱导的肺纤维化的类型、发展阶段和特征。此外,纤维化是一个动态而复杂的过程,再加上患者的异质性,即使由同一病毒引起,患者之间也可能存在很大差异。为了对病毒引起的肺纤维化进行更全面的评估,需要结合多种手段,如HRCT、病理方法和分子诊断。
综上所述,明确病毒性肺纤维化的临床症状、组织病理学和分子特征,并探索其潜在机制,对病毒引起的肺纤维化的早期诊断和治疗具有重要意义。
缩写
ACE2:血管紧张素转换酶 2
Ang II:血管紧张素 II
ARDS:急性呼吸窘迫综合征
ATL:成人T细胞白血病淋巴瘤
BALF:支气管肺泡灌洗液
CMV:巨细胞病毒
CTGF:结缔组织生长因子
DAD:弥漫性肺泡损伤
DPP4:二肽基肽酶4
EBV:爱泼斯坦-巴尔病毒
ECM:细胞外基质
EGF:表皮生长因子
EMT:上皮-间充质转化
FGF:成纤维细胞生长因子
HIV:人类免疫缺陷病毒
HTLV:人类嗜T淋巴细胞病毒
MCP-1:单核细胞化学引诱蛋白1
MERS:中东呼吸综合征
MHV68:鼠γ疱疹病毒 68
MMP:基质金属蛋白酶
PAI-1:纤溶酶原激活剂抑制剂-1
PDGF:血小板来源生长因子
RAS:肾素-血管紧张素系统
SARS:严重急性呼吸综合征
TGF-β:转化增长因子β
TIMP:金属蛋白酶的组织抑制剂
TNF-α:肿瘤坏死因子-α
参考文献 (可上下滑动浏览)
1. Reynolds HY. Lung infammation and fibrosis: an alveolar macrophage centered perspective from the 1970s to 1980s. Am J Respir Crit Care Med. 2005;171:98-102.
2. Han X, et al. Six-month follow-up chest CT findings after severe COVID-19 pneumonia. Radiology. 2021;299:E177-186.
3. Yamagishi M, et al. HTLV-1-mediated epigenetic pathway to adult T-cell leukemia-lymphoma. Front Microbiol. 2018;9:1686.
4. Yamashiro T, et al. CT scans of the chest in carriers of human T-cell lymphotropic virus type 1: presence of interstitial pneumonia. Acad Radiol. 2012;19:952-7.
5. Cachay R, et al. Clinical, radiological and functional characteristics of pulmonary diseases among HTLV-1 infected patients without prior active tuberculosis infection. Pathogens. 2021.
6. Kim SJ, et al. Transactivation of the transforming growth factor beta 1 (TGF-beta 1) gene by human T lymphotropic virus type 1 tax: a potential mechanism for the increased production of TGF-beta 1 in adult T cell leukemia. J Exp Med. 1990;172:121-9.
7. Teruya H, et al. Human T-cell leukemia virus type I infects human lung epithelial cells and induces gene expression of cytokines, chemokines and cell adhesion molecules. Retrovirology. 2008;5:86.
8. Matsuyama W, et al. Infuence of human T lymphotrophic virus type I on cryptogenic fbrosing alveolitis-HTLV-I associated fbrosing alveolitis: proposal of a new clinical entity. Clin Exp Immunol. 2003;133:397-403.
9. Miyazato A, et al. Chemokine synthesis and cellular infammatory changes in lungs of mice bearing p40tax of human T-lymphotropic virus type 1. Clin Exp Immunol. 2000;120:113-24.
10. Dofman SR, Miller RF. Interstitial lung disease in HIV. Clin Chest Med. 2013;34:293-306.
11. Crothers K, et al. HIV infection and risk for incident pulmonary diseases in the combination antiretroviral therapy era. Am J Respir Crit Care Med. 2011;183:388-95.
12. Leader JK, et al. Risk factors associated with quantitative evidence of lung emphysema and fbrosis in an HIV-infected cohort. J Acquir Immune Defc Syndr. 2016;71:420-7.
13. Marts LT, Guidot DM, Sueblinvong V. HIV-1 protein gp120 induces mouse lung fbroblast- to-myofbroblast transdiferentiation via CXCR4 activation. Am J Med Sci. 2019; 357:483-91.
14. Pellicelli AM, et al. Pathogenesis of HIV-related pulmonary hypertension.Ann N Y Acad Sci.
2001;946:82-94.
15. Sime PJ, et al. Transfer of tumor necrosis factor-alpha to rat lung inducessevere pulmonary infammation and patchy interstitial fbrogenesis with induction of transforming growth factor- beta1 and myofbroblasts. Am J Pathol. 1998;153:825-32.
16. Warshamana GS, Corti M, Brody AR. TNF-alpha, PDGF, and TGF-beta(1) expression by primary mouse bronchiolar-alveolar epithelial and mesenchymal cells: tnf-alpha induces TGF- beta(1). Exp Mol Pathol.2001;71:13-33.
17. Meng Y, et al. Angiotensin-converting enzyme 2/angiotensin-(1-7)/ Mas axis protects against lung fbrosis by inhibiting the MAPK/NF-κB pathway. Am J Respir Cell Mol Biol. 2014;50: 723-36.
18. Cook CH, et al. Pulmonary cytomegalovirus reactivation causes pathology in immuno- competent mice. Crit Care Med. 2006;34:842-9.
19. Cook CH, et al. Occult herpes family viral infections are endemic in critically ill surgical patients. Crit Care Med. 2003;31:1923-9.
20. Balthesen M, Messerle M, Reddehase MJ. Lungs are a major organ site of cytomegalovirus latency and recurrence. J Virol. 1993;67:5360-6.
21. Yonemaru M, et al. Elevation of antibodies to cytomegalovirus and other herpes viruses in pulmonary fbrosis. Eur Respir J. 1997;10:2040-5.
22. Franquet T, Lee KS, Müller NL. Thin-section CT fndings in 32 immunocompromised patients with cytomegalovirus pneumonia who do not have AIDS. AJR Am J Roentgenol. 2003;181: 1059-63.
23. Papazian L, et al. Cytomegalovirus. An unexpected cause of ventilator associated pneumonia. Anesthesiology. 1996;84:280-7.
24. Li Y, et al. Latent cytomegalovirus infection exacerbates experimental pulmonary fibrosis by activating TGF-beta1. Mol Med Rep. 2016;14:1297-301.
25. Walton KL, Johnson KE, Harrison CA. Targeting TGF-β mediated SMAD signaling for the prevention of fbrosis. Front Pharmacol. 2017;8:461.
26. She YX, Yu QY, Tang XX. Role of interleukins in the pathogenesis of pulmonary fbrosis. Cell Death Discov. 2021;7:52.
27. van de Berg PJ, et al. Cytomegalovirus infection reduces telomere length of the circulating T cell pool. J Immunol. 2010;184:3417-23.
28. Duckworth A, et al. Telomere length and risk of idiopathic pulmonary fbrosis and chronic obstructive pulmonary disease: a mendelian randomisation study. Lancet Respir Med. 2021;9: 285-94.
29. Ankermann T, et al. Chronic interstitial lung disease with lung fbrosis in a girl: uncommon sequelae of Epstein–Barr virus infection. Pediatr Pulmonol. 2003;35:234-8.
30. Malizia AP, et al. Alveolar epithelial cell injury with Epstein–Barr virus upregulates TGFbeta1 expression. Am J Physiol Lung Cell Mol Physiol. 2008;295:L451-60.
31. Sides MD, et al. The Epstein-Barr virus latent membrane protein 1 and transforming growth factor-beta1 synergistically induce epithelial–mesenchymal transition in lung epithelial cells. Am J Respir Cell Mol Biol. 2011;44:852-62.
32. Krug LT, et al. Inhibition of NF-kappaB signaling reduces virus load and gammaherpesvirus- induced pulmonary fbrosis. Am J Pathol.2010;177:608-21.
33. Mora AL, et al. Lung infection with gamma-herpesvirus induces progressive pulmonary fbrosis in Th2-biased mice. Am J Physiol Lung Cell Mol Physiol. 2005;289:L711-21.
34. Mora AL, et al. Control of virus reactivation arrests pulmonary herpesvirus-induced fbrosis in IFN-gamma receptor-defcient mice. Am J Respir Crit Care Med. 2007;175:1139-50.
35. Shieh WJ, et al. 2009 pandemic infuenza A (H1N1): pathology and pathogenesis of 100 fatal cases in the United States. Am J Pathol. 2010;177:166-75.
36. Nakajima N, et al. Histopathological and immunohistochemical fndings of 20 autopsy cases with 2009 H1N1 virus infection. Mod Pathol. 2012;25:1-13.
37. Li P, et al. Serial evaluation of high-resolution CT fndings in patients with pneumonia in novel swine-origin infuenza A (H1N1) virus infection. Br J Radiol. 2012;85(1014):729-35.
38. Mineo G, et al. Post-ARDS pulmonary fbrosis in patients with H1N1 pneumonia: role of follow-up CT. Radiol Med. 2012;117:185-200.
39. Saha A, et al. Combined pirfenidone, azithromycin and prednisolone in post-H1N1 ARDS pulmonary fbrosis. Sarcoidosis Vasc Difuse Lung Dis.2018;35:85-90.
40. Wen Y, et al. Immunological features in patients with pneumonitis due to infuenza A H1N1 infection. J Investig Allergol Clin Immunol. 2011;21:44-50.
41. Shatskaya EV, et al. Study of SMAD-dependent signal pathway in the development of early pulmonary fbrosis in mice infected with infuenza A/H1N1 virus. Bull Exp Biol Med. 2017;162:647-9.
42. Drakopanagiotakis F, et al. Apoptosis in lung injury and fbrosis. Eur Respir J. 2008; 32: 1631
-8.
43. Roberson EC, et al. Infuenza induces endoplasmic reticulum stress, caspase-12-dependent apoptosis, and c-Jun N-terminal kinase-mediated transforming growth factor-β release in lung epithelial cells. Am J Respir Cell Mol Biol. 2012;46:573-81.
44. Lu PX, et al. Radiological features of lung changes caused by avian infuenza subtype A H5N1 virus: report of two severe adult cases with regular follow-up. Chin Med J. 2010;123:100-4.
45. Chen C, Chen J, Huang JA. Persistence of lymphocytopenia with CT abnormalities among patients with critical H7N9 swine-origin infuenza A virus infection. Jpn J Radiol. 2015;33: 657 -62.
46. Gao HN, et al. Clinical fndings in 111 cases of infuenza A (H7N9) virus infection. N Engl J Med. 2013;368:2277-85.
47. To KF, et al. Pathology of fatal human infection associated with avian infuenza A H5N1 virus. J Med Virol. 2001;63:242-6.
48. Huang JB, et al. Histopathological fndings in a critically ill patient with avian infuenza A (H7N9). J Thorac Dis. 2015;7:E672-7.
49. Qiao J, et al. Pulmonary fbrosis induced by H5N1 viral infection in mice. Respir Res. 2009; 10:107.
50. Xu T, et al. Acute respiratory distress syndrome induced by avian infuenza A (H5N1) virus in mice. Am J Respir Crit Care Med.2006;174:1011-7.
51. Anikina AG, et al. Expression of profbrotic growth factors and their receptors by mouse lung macrophages and fbroblasts under conditions of acute viral infammation in infuenza A/H5N1 virus. Bull Exp Biol Med. 2014;156:833-7.
52. Zou Z, et al. Angiotensin-converting enzyme 2 protects from lethal avian infuenza A H5N1 infections. Nat Commun. 2014;5:3594.
53. Huang F, et al. Angiotensin II plasma levels are linked to disease severity and predict fatal outcomes in H7N9-infected patients. Nat Commun. 2014;5:3595.
54. Meng Y, et al. The angiotensin-converting enzyme 2/angiotensin (1–7)/ Mas axis protects against lung fbroblast migration and lung fbrosis by inhibiting the NOX4-derived ROS-mediated RhoA/Rho kinase pathway. Antioxid Redox Signal. 2015;22:241-58.
55. Grillo F, et al. Lung fbrosis: an undervalued fnding in COVID-19 pathological series. Lancet Infect Dis. 2021;21:e72.
56. Kuba K, et al. Lessons from SARS: control of acute lung failure by the SARS receptor ACE2. J Mol Med. 2006;84:814-20.
57. Arabi YM, et al. Middle East respiratory syndrome. N Engl J Med. 2017;376:584-94.
58. Ng DL, et al. Clinicopathologic, immunohistochemical, and ultrastructural fndings of a fatal case of middle east respiratory syndrome coronavirus infection in the United Arab Emirates, April 2014. Am J Pathol. 2016;186:652-8.
59. Das KM, et al. Follow-up chest radiographic fndings in patients with MERS-CoV after recovery. Indian J Radiol Imaging. 2017;27:342-9.
60. Zhou J, et al. Active replication of Middle East respiratory syndrome coronavirus and aberrant induction of infammatory cytokines and chemokines in human macrophages: implications for pathogenesis. J Infect Dis. 2014;209:1331-42.
61. Zielecki F, et al. Human cell tropism and innate immune system interactions of human respiratory coronavirus EMC compared to those of severe acute respiratory syndrome coronavirus. J Virol. 2013;87:5300-4.
62. Yeung ML, et al. MERS coronavirus induces apoptosis in kidney and lung by upregulating Smad7 and FGF2. Nat Microbiol. 2016;1:16004.
63. Kim J, et al. Middle East respiratory syndrome-coronavirus infection into established hDPP4-transgenic mice accelerates lung damage via activation of the pro-infammatory response and pulmonary fibrosis. J Microbiol Biotechnol. 2020;30:427-38.
64. Falzarano D, et al. Infection with MERS-CoV causes lethal pneumonia in the common marmoset. PLoS Pathog. 2014;10: e1004250.
65. Stadler K, et al. SARS-beginning to understand a new virus. Nat Rev Microbiol. 2003; 1: 209-18.
66. Tsui PT, et al. Severe acute respiratory syndrome: clinical outcome and prognostic correlates. Emerg Infect Dis. 2003;9:1064-9.
67. Gu J, Korteweg C. Pathology and pathogenesis of severe acute respiratory syndrome. Am J Pathol. 2007;170:1136-47.
68. Hwang DM, et al. Pulmonary pathology of severe acute respiratory syndrome in Toronto. Mod Pathol. 2005;18:1-10.
69. Xie L, et al. Follow-up study on pulmonary function and lung radiographic changes in rehabilitating severe acute respiratory syndrome patients after discharge. Chest. 2005; 127:2119-24.
70. Hui DS, et al. Impact of severe acute respiratory syndrome (SARS) on pulmonary function, functional capacity and quality of life in a cohort of survivors. Thorax. 2005;60:401-9.
71. Zhang P, et al. Long-term bone and lung consequences associated with hospital-acquired severe acute respiratory syndrome: a 15-year followup from a prospective cohort study. Bone Res. 2020;8:8.
72. Xie L, et al. Dynamic changes of serum SARS-coronavirus IgG, pulmonary function and radiography in patients recovering from SARS after hospital discharge. Respir Res. 2005;6:5.
73. Rockx B, et al. Early upregulation of acute respiratory distress syndrome associated cytokines promotes lethal disease in an aged-mouse model of severe acute respiratory syndrome coronavirus infection. J Virol. 2009;83:7062-74.
74. Zhao X, Nicholls JM, Chen YG. Severe acute respiratory syndromeassociated coronavirus nucleocapsid protein interacts with Smad3 and modulates transforming growth factor-beta signaling. J Biol Chem. 2008;283:3272-80.
75. Gralinski LE, et al. Mechanisms of severe acute respiratory syndrome coronavirus-induced acute lung injury. Bio. 2013.
76. Courey AJ, et al. The vitronectin-binding function of PAI-1 exacerbates lung fbrosis in mice. Blood. 2011;118:2313-21.
77. Rana T, et al. PAI-1 regulation of TGF-β1-induced alveolar type II cell senescence, SASP secretion, and SASP-mediated activation of alveolar macrophages. Am J Respir Cell Mol Biol. 2020;62:319-30.
78. Page C, et al. Induction of alternatively activated macrophages enhances pathogenesis during severe acute respiratory syndrome coronavirus infection. J Virol. 2012;86:13334-49.
79. Gieseck RL 3rd, Wilson MS, Wynn TA. Type 2 immunity in tissue repair and fbrosis. Nat Rev Immunol. 2018;18:62-76.
80.Grillo F, et al. Lung fbrosis: an undervalued fnding in COVID-19 pathological series. Lancet Infect Dis. 2021;21:e72.
81. Guan WJ, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382:1708-20.
82. Yu M, et al. Prediction of the development of pulmonary fbrosis using serial thin-section CT and clinical features in patients discharged after treatment for COVID-19 pneumonia. Korean J Radiol. 2020;21:746–55.
83. Mo X, et al. Abnormal pulmonary function in COVID-19 patients at time of hospital discharge. Eur Respir J, 2020:1217-2020.
84. Lax SF, et al. Pulmonary arterial thrombosis in COVID-19 with fatal out come : results from a prospective, single-center clinicopathologic case series. Ann Intern Med. 2020;173:350-61.
85. Schwensen HF, et al. Fatal pulmonary fbrosis: a post-COVID-19 autopsy case. J Clin Pathol. 2020.206879.
86. Li X, et al. Pulmonary fbrosis and its related factors in discharged patients with new corona virus pneumonia: a cohort study. Respir Res. 2021;22:203.
87. Pan Y, et al. Initial CT fndings and temporal changes in patients with the novel coronavirus pneumonia (2019-nCoV): a study of 63 patients in Wuhan, China. Eur Radiol. 2020;30:3306-9.
88. Zhang H, et al. Histopathologic changes and SARS-CoV-2 immunostaining in the lung of a patient with COVID-19. Ann Intern Med. 2020;172:629-32.
89. Hu ZJ, et al. Lower circulating interferon-gamma is a risk factor for lung fbrosis in COVID-19 patients. Front Immunol. 2020;11: 585647.
90. Valdebenito S, et al. COVID-19 lung pathogenesis in SARS-CoV-2 autopsy cases. Front Immunol. 2021;12: 735922.
91. Munster VJ, et al. Respiratory disease in rhesus macaques inoculated with SARS-CoV-2. Nature. 2020;585:268-72.
92. Shan C, et al. Infection with novel coronavirus (SARS-CoV-2) causes pneumonia in Rhesus macaques. Cell Res. 2020;30:670-7.
93. Harrison AG, Lin T, Wang P. Mechanisms of SARS-CoV-2 transmission and pathogenesis. Trends Immunol. 2020;41:1100-15.
94. Zou H, Li SQ. Pulmonary fbrosis in critically ill patients with novel coronavirus pneumonia during the convalescent stage and a proposal for early intervention. Acta Pharmacol Sin. 2020.
95. Krishna Murthy P, et al. Repurposing of histone deacetylase inhibitors: a promising strategy to combat pulmonary fbrosis promoted by TGF beta signalling in COVID-19 survivors. Life Sci. 2021;266: 118883.
96. Chen G, et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest. 2020;130:2620-9.
97. Pedroza M, et al. STAT-3 contributes to pulmonary fbrosis through epithelial injury and fbroblast-myofbroblast diferentiation. FASEB J. 2016;30:129-40.
98. Xu J, et al. SARS-CoV-2 induces transcriptional signatures in human lung epithelial cells that promote lung fbrosis. Respir Res. 2020;21:182.
99. Leng L, et al. Pathological features of COVID-19-associated lung injury: a preliminary proteomics report based on clinical samples. Signal Transduct Target Ther. 2020;5:240.
100. Crisan-Dabija R, et al. “A chain only as strong as its weakest link”: an upto-date literature review on the bidirectional interaction of pulmonary fbrosis and COVID-19. J Proteome Res. 2020;19:4327-38.
101. Chen X, et al. Coronavirus membrane-associated papain-like proteases induce autophagy through interacting with Beclin1 to negatively regulate antiviral innate immunity. Protein Cell. 2014;5:912-27.
102. Yim WW, Mizushima N. Autophagosome maturation stymied by SARSCoV-2. Dev Cell. 2021;56:400-2.
103. Campbell GR, et al. Human immunodefciency virus type 1 Nef inhibits autophagy through transcription factor EB sequestration. PLoS Pathog. 2015;11: e1005018.
104. Chaumorcel M, et al. The human cytomegalovirus protein TRS1 inhibits autophagy via its interaction with Beclin 1. J Virol. 2012;86:2571-84.
105. Mouna L, et al. Analysis of the role of autophagy inhibition by two complementary human cytomegalovirus BECN1/Beclin 1-binding proteins. Autophagy. 2016;12:327-42.
106. Ramanathan A, et al. Transient unfolding and long-range interactions in viral BCL2 M11 enable binding to the BECN1 BH3 domain. Biomolecules. 2020.
107. Hill C, et al. Autophagy inhibition-mediated epithelial-mesenchymal transition augments local myofbroblast diferentiation in pulmonary fbrosis. Cell Death Dis. 2019;10:591.
108. Zhao H, et al. Autophagy, an important therapeutic target for pulmonary fbrosis diseases. Clin Chim Acta. 2020;502:139-47.
109. Diem K, et al. Mechanical stretch activates piezo1 in caveolae of alveolar type I cells to trigger ATP release and paracrine stimulation of surfactant secretion from alveolar type II cells. Faseb J. 2020;34:12785-804.












译者简介

章凯毅
安徽中医药⾼等专科学校2021级规培学员(临床医学系,预防医学)。

李超
呼吸内科主任医师,芜湖市第一人民医院呼吸二科兼感染科副主任,安徽省预防医学会呼吸病预防与控制专业委员会委员,安徽省抗癌协会皖西南肺癌专委会委员,主要专业方向为呼吸系统感染和胸部肿瘤。

胥武剑
博士、副主任医师、博士后;上海市东方医院呼吸与危重症医学科 副主任医师;同济大学医学院硕士生导师;上海市医学会介入学组 委员、秘书;中国老年保健医学研究会呼吸病学分会 委员;中国防痨协会MDT专业分会 委员;中国人体健康科技促进会呼吸介入专委会 常委;擅长肺癌的规范化诊疗,肺部肿瘤的介入诊疗,危重肺部感染的诊断和治疗;近年来先后获国家自然科学基金、上海市科委、中国博士后、江苏省博士后等各项基金6项,发表SCI论文11篇,获国家专利11项。
* 文章仅供医疗卫生相关从业者阅读参考
本文完
责编:Jerry