

 分享
分享

目的:通过分析总结肺毛细血管瘤病的临床特征,提高临床医生对肺毛细血管瘤病的认识及诊疗水平。
方法:回顾并分析南京鼓楼医院呼吸与危重症医学科收治的2例肺毛细血管瘤病患者的临床表现、影像学特征及组织病理学等资料;同时以“肺毛细血管瘤病”和“Pulmonary Capillary Hemangiomatosis”为关键词,在国内外数据库共检索到肺毛细血管瘤病相关文献227篇,对其中113篇肺毛细血管瘤病相关病例报告进行分析。
结果:本文报道了2例肺毛细血管瘤病,患者均先被误诊为其他疾病,跟踪随访发现治疗效果不佳,后通过外科肺活检确诊。例1患者确诊4个月后行肺移植手术,例2患者规律服用降肺动脉压药物,目前2例患者症状稳定,肺动脉压力明显降低。在文献复习中,93例患者病历资料进入本次回顾性研究,对这些患者的确诊手段、临床表现、基因检测、检查结果、影像学特征、病理诊断和生存预后等进行了归纳总结。
结论:肺毛细血管瘤病在临床中是一个罕见病,其发病率极低,临床表现大多无特异性,极易误诊、漏诊,充分认识并掌握其临床特征是未来降低漏诊、误诊的关键。
肺毛细血管瘤病(pulmonary capillary hemangiomatosis,PCH)是一种极为罕见、预后不良的肺血管增殖性疾病,以肺泡壁和肺泡间隔的毛细血管异常增生为病理特征,肺血管阻力进行性升高,最终导致无法逆转的肺动脉高压和右心衰竭[1],因此早期准确识别该疾病、进行有效干预是临床诊治中的关键。本研究通过回顾并分析南京鼓楼医院呼吸与危重症医学科收治的2例PCH患者的临床表现、影像学特征及组织病理学等资料,以期提高临床医生对肺毛细血管瘤病的认识及诊疗水平。
病例资料
例1
患者女,39岁,因“活动后气喘1年余,加重3个月”于2022年5月11日收入我科。患者于2021年初起无明显诱因下出现活动后气喘,偶伴轻微干咳,无畏寒发热,无胸痛咯血。入院前3个月患者至外院就诊,查胸部CT示两肺见弥漫多发磨玻璃样密度影;肺功能检查示中度限制性通气功能障碍,肺一氧化碳弥散量(diffusing capacity of the lungs for carbon monoxide,DLCO)<40%预计值,呈重度肺弥散功能障碍;超声心动图示右心增大,肺动脉增宽,估测肺动脉收缩压为45 mm Hg(1 mm Hg=0.133 kPa)(轻度肺动脉高压)。数天后患者行支气管镜检查,支气管肺泡灌洗液(bronchoalveolar lavage fluid,BALF)细胞学检查:巨噬细胞占60%,淋巴细胞占40%,未见抗酸杆菌、革兰阳性球菌、革兰阴性杆菌和真菌孢子及假丝;经支气管镜肺活检术(transbronchial lung biopsy,TBLB)病理:送检肺组织局灶区肺泡间隔增宽伴间质少量慢性炎细胞浸润及碳末沉着,未见异型细胞。综合以上考虑过敏性肺炎,给予糖皮质激素抗炎治疗3个月。入院前1周复查胸部CT仍示两肺弥漫性病变,较前无明显吸收,且症状较前加重,遂为求进一步诊治收住入院。既往无吸烟史、无化学性物质、毒品接触史,长期经营艾灸香薰工作。
入院查体:T 36.8℃,P 70次/min,R 20次/min,BP 97/70 mm Hg,神清,口唇无发绀,全身浅表淋巴结未及肿大,颈静脉无充盈,肝颈静脉回流征阴性,胸廓无畸形,双侧呼吸运动对称,双肺呼吸音粗,未闻及及捻发音;心界不大,心率70次/min,律齐,心脏听诊P2亢进;腹部平软,肝、脾肋下未触及,双下肢无水肿。
入院后完善相关检查。血气分析:pH 7.443,PCO2 26.3 mm Hg,PO2 71.0 mm Hg,SpO2 95%(未吸氧),氧合指数(oxygenation index,OI)338 mm Hg。血常规、生化全套、D-二聚体正常,结核感染T细胞检测、血GM/G试验、EB病毒/巨细胞病毒正常。p-ANCA弱阳性。超声心动图:三尖瓣中-重度反流,肺动脉内径增宽,估测肺动脉收缩压103 mm Hg(重度肺动脉高压);心肌运动瓣环位移分析提示右心功能下降,瓣膜重度反流。CT肺动脉造影(computed tomographic pulmonary angiography,CTPA)(图1):肺动脉主干轻度增粗,管径约3.4 cm,未见充盈缺损征象,两肺野纹理增粗,两肺可见多发小片及结节状磨玻璃密度影。考虑此前支气管镜检查的风险及结果,患者权衡了经支气管镜冷冻肺活检和外科胸腔镜肺活检的利弊,要求直接进行外科肺活检术。
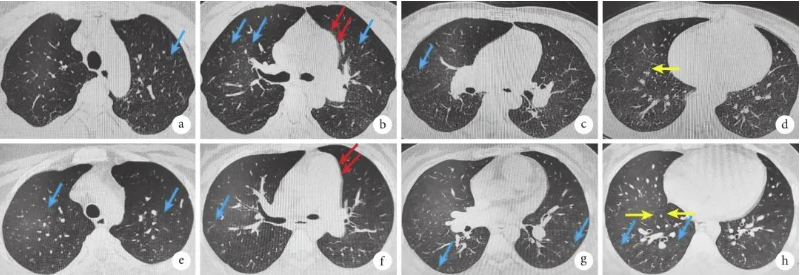
图1 例1(a~d)和例2(e~h)患者的胸部CT像
均可见呈小叶中心分布的多发磨玻璃影、小叶间隔增厚以及肺动脉主干增宽。其中,磨玻璃结节(蓝箭)、小叶间隔增厚(黄箭)、肺动脉主干增宽(红箭)。
5月18日进行外科胸腔镜下左肺楔形切除术。术后病理(图2):送检肺组织多数区域示肺气肿样改变,广泛肺淤血,散在多灶肺泡隔增厚、毛细血管增多,肺实质内见肌化细小动脉,小气道旁动脉扩张、充血,组织学符合肺毛细血管瘤病、肺气肿及肺动脉高压;免疫组化示CD31(+)、CD34(+)、CK7(+),特殊染色示弹力纤维染色(+)、网状纤维染色(+)、Van Gieson染色(+);基因检测示EIF2AK4双等位基因突变。患者PCH诊断明确,予西地那非改善肺动脉高压、呋塞米联合螺内酯利尿等治疗后,患者症状好转后出院。同年9月患者于外院行双肺移植术,现定期复查心超示肺动脉压力正常,目前仍在随访中。
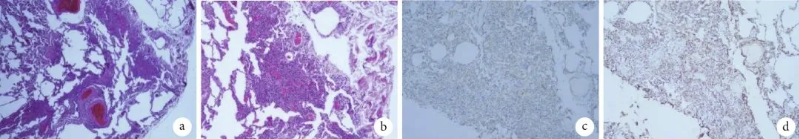
图2 例1患者胸腔镜下左肺楔形切除术术后病理像
a~b. 肺泡壁和肺泡间隔的毛细血管广泛增生,肺间质可见肺小动脉代偿性肌化(HE×40,HE×100);c. CD31阳性表达(免疫组化×100);d. CD34阳性表达(免疫组化×100)。
例2
患者女,27岁,因“间断咯血4个月余,胸闷气喘3个月”于2022年11月30日收入我科。入院前4个月患者无明显诱因下出现咯血3口,色鲜红,量约10 mL,无牙龈出血、黑便,立即至外院就诊。血常规:白细胞计数14.48×109/L,中性粒细胞计数12.13×109/L,红细胞计数3.77×1012/L,血红蛋白84 g/L;胸部CT:两肺弥漫性病变,考虑感染可能;给予左氧氟沙星0.5 g qd抗感染、氨甲环酸2 g qd止血、甲泼尼龙80 mg qd抗炎治疗,咯血停止。入院前3个月患者出现活动后胸闷气喘等不适,未重视,入院前再次咯血,量约20 mL,遂为求进一步诊治收住入院。既往无吸烟史、无职业粉尘等接触史,从事内科护理工作。
入院查体:T 36.5℃,P 75次/min,R 20次/min,BP 110/61 mm Hg,神清,口唇无发绀,全身浅表淋巴结未及肿大,颈静脉无充盈,肝颈静脉回流征阴性,胸廓无畸形,双肺呼吸音粗,触觉语颤对称,两下肺未闻及皮下捻发音;心界不大,心率75次/min,律齐,心脏听诊P2亢进;腹部平软,肝、脾肋下未触及,双下肢无浮肿。
入院后完善相关检查。血气分析:pH 7.409,PCO2 32.6 mm Hg,PO2 125 mm Hg,SpO2 96%(鼻导管吸氧3 L/min),OI 431 mm Hg。血常规:白细胞计数27.1×109/L,红细胞计数4.52×1012/L,血红蛋白98 g/L,中性粒细胞计数20.9×109/L,血小板计数533×109/L。类风湿三项、自身抗体、抗中性粒细胞胞浆抗体、自身免疫性肌炎抗体正常,呼吸道感染病原体阴性。心电图:窦性心律,PR间期缩短、V1导联R/S>1;胸部CT:两肺弥漫性模糊影,两肺小结节,肺动脉主干轻度增粗(图1)。超声心动图:二尖瓣、三尖瓣轻度反流,肺动脉内径增宽,估测肺动脉收缩压47 mm Hg(轻-中度肺动脉高压)。复查患者血常规示三系较前变化不大,考虑血液系统疾病不排除,行骨髓穿刺+活检术:缺铁性贫血,骨髓感染像。进一步完善支气管镜和TBLB检查:送检少量肺组织示肺泡腔出血,未见肿瘤细胞,未见明确血管炎症等病变。
诊断考虑弥漫性肺泡出血综合征(特发性),给予左氧氟沙星抗感染、甲泼尼龙抗炎等综合治疗,无咯血后出院。出院1个月后复查胸部CT较前稍有吸收趋势,嘱咐继续激素口服治疗。2023年3月中旬患者爬楼时仍感胸闷气喘,伴头晕,至外院查超声心动图示肺动脉收缩压150 mm Hg,复查胸部CT示病灶较前无明显吸收,再次至我院就诊。经与患者本人及家属商议,考虑到前期治疗效果不佳,且肺上同时存在高危结节,动员进行外科肺活检术。3月17日在全麻下经外科胸腔镜行右下肺部分切除术,术后病理:送检肺组织部分区域肺泡上皮增生,倾向非粘液性微浸润性腺癌;免疫组化示1号蜡块:腺上皮表达TTF-1(+),Ki67(约5%+),P40(+),CK5/6(小灶+),P63(+),血管壁平滑肌肌动蛋白(smooth muscle actin,SMA)(–),Calponin(–),结合免疫组化染色结果考虑非黏液性原位腺癌伴微小浸润;部分区域肺泡塌陷、肺泡隔毛细血管增多,组织学倾向毛细血管瘤病;免疫组化(5号、8号蜡块):CD31、CD34显示血管,CK(腺上皮+),结合免疫组化染色结果符合肺毛细血管瘤病。基因检测未见EIF2AK4双等位基因突变。最终,患者明确诊断为PCH。予安生立坦降压、西地那非改善肺动脉高压、呋塞米联合螺内酯利尿等治疗,症状好转出院后继续用药,定期至我院复查。目前患者活动耐量较前增加,最近一次复查心超肺动脉收缩压预估为35 mm Hg左右,我科继续随诊。
2、文献复习及总结
在知网、万方国内数据库以“肺毛细血管瘤病”为检索词对2024年4月之前的文献进行检索,有关肺毛细血管瘤病的文献共22篇;在PubMed等国外数据库以“Pulmonary Capillary Hemangiomatosis”为检索词对2024年4月之前的文献进行检索,有关PCH的文献共205篇。经过删除综述、病例重复报告、资料不全或最终诊断不确定等文献,共纳入113篇文献,含148例病例。结合上述2例患者既往身体健康、无慢性病史等,考虑到合并或继发于其他疾病时无法判断PCH为单发性疾病还是反应性增生的结果,设立如下排除标准:(1)孤立性肺毛细血管瘤(solitary pulmonary capillary hemangioma,SPCH)或病理特征为PCH样病灶;(2)PCH继发于结缔组织病(connective tissue disease,CTD)或免疫系统性疾病,如系统性硬化症、关节炎、系统性红斑狼疮等,或患者存在房/室间隔缺损/狭窄的先天性心脏病,或遗传性毛细血管扩张症(hereditary hemorrhagic telangiectasia,HHT)等涉及内脏损害的疾病;(3)药物诱导发生的PCH;(4)患者发病年龄<2岁。最终纳入93例资料完整的病例,对其中患者的确诊手段、临床表现、基因检测、肺功能和血流动力学指标、影像学特征、病理诊断和生存预后等进行归纳总结。排除病例:SPCH或病理特征为PCH样病灶22例;PCH继发于CTD或免疫系统性疾病11例,如系统性硬化症、关节炎、系统性红斑狼疮等,或患者存在房/室间隔缺损/狭窄的先天性心脏病4例,或HHT等涉及内脏损害的疾病1例;药物诱导发生PCH的1例;患者发病年龄<2岁的16例。最终共研究93例,这些病例均结合临床表现、基因检测、肺功能和血流动力学指标、影像学特征和病理诊断等确诊。
2.1 确诊时间段和确诊方式
生前确诊69例,尸检确诊24例。生前确诊的患者中:(1)结合临床表现、肺功能和血流动力学指标、影像学和组织病理确诊57例,其中4例患者同时完善基因检测,结果示阳性;(2)结合典型的临床症状、肺功能和血流动力学指标、特征性的影像学表现确诊6例;(3)结合基因检测阳性、血流动力学指标和影像学表现确诊6例。
2.2 人口学特征
男性患者49例,女性患者43例,信息未知(性别、年龄)1例,平均发病年龄36岁(范围为7~99岁)。
2.3 临床表现
以劳力性呼吸困难为首发症状者88例,其他首发症状尚有咯血4例,外周水肿1例。所有患者中,出现外周水肿11例,出现咯血症状9例,出现劳力性晕厥发作6例,肺部听诊闻及湿啰音5例,出现杵状指3例。
2.4 可能的病因和发病机制
完成基因检测14例,存在EIF2AK4双等位基因突变10例,不存在该基因突变4例。职业相关环境暴露2例,与有机溶剂相关。
2.5 肺功能和血流动力学指标
完善肺功能检查42例,完善血气分析26例,完善超声心动图53例,完善右心导管检查50例,完善运动耐量评估17例,完善核素肺通气/灌注显像(ventilation/perfusion,V/Q)15例。
2.6 影像学特征
完善X线检查47例,绝大多数患者X线均可见肺动脉段凸出,有12例患者的X线呈现双侧间质浸润伴肺动脉扩张的改变。完善胸部CT检查72例,PCH的高分辨率CT(high-resolution computed tomography,HRCT)表现包括小叶中心磨玻璃阴影、胸膜下间隔线增厚、纵隔淋巴结肿大等,以弥漫多发磨玻璃影,呈小叶中心分布最为常见,其中仅表现出多发磨玻璃阴影48例,表现为弥漫多发磨玻璃影和胸膜下间隔线增厚两联征的10例,表现出弥漫多发磨玻璃影和纵隔淋巴结肿大两联征的6例,同时可见小叶中心磨玻璃阴影、胸膜下间隔线增厚和纵隔淋巴结肿大这三种征象的5例,CT呈非特异性改变3例。
2.7 支气管镜检查
完善支气管镜检查11例(11/93),其中BALF中可见含铁血黄素细胞3例,经支气管冷冻肺活检获得组织学标本明确病理诊断2例,通过TBLB取得组织明确病理检查1例,肺组织呈非特异性慢性炎症2例,BALF提示肺泡出血1例,BALF内可见大量细菌1例,送检肺组织可见肺泡隔纤维化伴细支气管化生1例。
2.8 病理检查
完善组织病理学检查81例。其中,肺组织活检57例,完善尸检24例。患者的肺泡间隙可见具有含铁血黄素的巨噬细胞25例。
2.9 随访
患者在随访阶段内仍存活32例,患者在随访阶段内已被告知死亡54例,患者失访7例(7/93)。死亡的54例患者大多因病情进展从而发生无法逆转的呼吸衰竭和心力衰竭。
2.10 肺移植
患者行肺移植手术22例,其中患者移植术后情况良好18例(18/22),其余4例患者分别死于移植术中、移植术后感染、移植术后PCH复发和术后并发症闭塞性细支气管炎。
3、讨论
3.1 概述
根据2022年欧洲心脏病学会/欧洲呼吸学会肺动脉高压诊疗指南(简称ESC/ERS指南),PCH作为一类具有明显肺毛细血管受累特征的疾病,目前被归类到动脉性肺动脉高压(pulmonary arterial hypertension,PAH)的亚组中[2]。PCH的发病年龄为2~70岁,可见于各个年龄段的人群,发病率无明显性别差异。PCH的发病机制尚未完全明确,近年研究发现家族性PCH的发病与EIF2AK4双等位基因突变密切相关,呈常染色体隐性遗传[3]。散发性PCH考虑与多种因素相关,如环境暴露包括烷化剂(环磷酰胺、丝裂霉素等)、有机溶剂(三氯乙烯等)和放射治疗等[4],或患者合并其他自身免疫性疾病(系统性硬化症、结节病、桥本甲状腺炎等)[5];部分散发性PCH患者也可能检测出上述基因突变。我们的2例患者均为女性,例1患者发病年龄为39岁,EIF2AK4基因突变阳性,例2患者发病年龄为27岁,EIF2AK4基因突变阴性,工作环境均无危险因素暴露,也未合并其他自身免疫性疾病。文献回顾中,男性患者49例,女性患者43例,信息未知(性别、年龄)1例,平均发病年龄36岁(范围7~99岁)。职业相关环境暴露2例,均与有机溶剂相关。
3.2 临床特征
3.2.1 临床表现
PCH起病多隐匿,常见症状包括非特异性的劳力性呼吸困难、咳嗽、乏力、胸闷和劳力性晕厥等,30%的患者可能出现咯血症状[6]。随着疾病进展,以上症状呈现进行性加重趋势。常见体征为肺动脉高压的表现,如心脏听诊可闻及肺动脉瓣区第二心音(P2)亢进,三尖瓣听诊区全收缩期吹风样杂音,胸骨左下缘收缩期可见抬举样搏动。体格检查时可见颈静脉怒张、外周水肿、腹腔积液或肝脏肿大等,提示右心衰竭。此外,查体时部分患者可见杵状指,肺部听诊可能闻及湿啰音。我们的2例患者均表现出活动后胸闷气喘,进行性呼吸困难等不适,心脏听诊均存在P2亢进,其中例2患者发病初期以间断咯血为首发症状。文献回顾中大部分患者以劳力性呼吸困难为首发和主要症状,休息后能缓解,进行性加重,这与我们的2例患者发病过程是一致的。上述PCH的症状和体征也可见于其他常见疾病,因此明确PCH诊断时患者可能已经出现无法逆转的肺动脉高压和右心衰竭等表现。
3.2.2 心电图表现
典型心电图表现为右心室肥厚、右心房增大、电轴右偏和右束支传导阻滞,具体包括V1导联R/S>1、肺性P波和QRS电轴右偏(≥+90°)等。部分患者心电图仅表现为非特异性QTc间期延长,少部分患者心电图甚至未见异常。因此,心电图只能对PCH的诊断起到线索和提示作用,并不能直接反映肺动脉压增高,用于PCH诊断的敏感性和特异性低,心电图正常并不能排除PCH。我们的例1患者心电图呈非特异性改变,例2患者心电图结果呈典型右心室肥厚表现。文献回顾中心电图异常16例,均提示右心室肥厚。
3.2.3 超声心动图
超声心动图是筛查肺动脉高压最重要的无创检查方法,可重复测量PCH患者的肺动脉内径宽度和肺动脉收缩压大小,同时还有助于排除左心相关性疾病。我们的2例患者在住院期间和随诊复查时均通过多次复查超声心动图来评价病情进展和治疗效果。文献回顾中完善超声心动图53例。超声心动图对评价心脏功能具有较好的价值,特别适合跟踪、随访,建议条件允许的情况下在同一家医院、由同一个医生进行随访检测更为适宜。
3.2.4 肺功能、动脉血气分析和运动耐量评估
肺功能检查是测量肺容量大小、检测气流有无受限的客观指标,对早期诊断肺部疾病、判断通气障碍类型具有重要的临床价值。PCH患者肺总量和用力肺活量通常在正常范围内,但DLCO严重降低,通常低于50%预计值[7]。我们的2例患者肺功能检查结果均提示DLCO严重降低。文献回顾中完善肺功能检查42例,所有患者均存在DLCO降低,呈中度至重度弥散功能障碍。
动脉血气分析是判断机体是否存在酸碱平衡紊乱,有无缺氧以及缺氧程度的可靠指标。PCH患者血气分析示动脉血氧分压下降,提示低氧血症[8]。我们的2例患者和文献回顾中的患者在疾病诊断阶段多次完善血气分析,结果提示进行性恶化低氧血症,随着病情发展,部分患者需要氧疗来保证全身氧气供给。
世界卫生组织(world health organization,WHO)肺动脉高压功能分级(functional classification,FC)、6分钟步行试验(6-minute walk test,6MWT)和心肺运动试验(cardiopulmonary exercise testing,CPET)是一些能够客观且无创地测定患者运动耐量的检查方法。用于评估肺动脉高压患者临床症状等级的WHO-FC,相较于纽约心脏病学会提出的NYHA心功能分级增加了对晕厥的描述[9]。大多数PCH患者在确诊时WHO肺动脉高压功能分级达到Ⅲ级或以上。6MWT这项检查实用易操作,结合患者步行距离大小和Borg自觉疲劳评分量表直观评估患者心肺功能。PCH患者在进行6MWT时血氧饱和度显著下降。CPET通过定量相关指标,可阐明运动不耐受、运动时相关症状的病理生理学机制。可惜的是,我们的2例患者诊治阶段均未行6MWT和CPET等运动耐量评估,文献回顾中提及完善运动耐量评估也仅17例。综上,临床工作中我们需要运用这些简单、无创的评估工具帮助判断治疗效果。
3.2.5 影像学检查
胸部X线片对PCH的诊断意义不大,可有肺动脉高压的X线征象,即肺动脉段凸出和右心室肥大,疾病迅速进展时可出现肺水肿。胸部HRCT是PCH患者的首选影像学检查方法。PCH在胸部CT上的表现以弥漫多发磨玻璃影,呈小叶中心分布最为常见,胸膜下间隔线增厚和纵隔淋巴结肿大这两种征象出现频率不高[10]。我们的2例患者胸部CT均可见呈小叶中心分布的多发磨玻璃影、小叶间隔增厚以及肺动脉主干增宽。文献回顾中,绝大多数患者X线均可见肺动脉段凸出,少部分患者可见双侧间质浸润伴中央肺动脉扩张;患者胸部CT同时可见小叶中心磨玻璃阴影、胸膜下间隔线增厚和纵隔淋巴结肿大这三种征象者仅有5例。因此,若患者仅具备弥漫多发小叶中心磨玻璃阴影的HRCT征象,在诊断意识淡薄的情况下,作为罕少见病的PCH恐难位列于鉴别诊断之中。
3.2.6 核素V/Q显像
核素V/Q显像是通过对比通气和相应的灌注图像,来识别影响肺功能的心肺疾病,还可以量化缺损程度。V/Q扫描敏感性高但特异性低,任何堵塞肺血管血流但无相应通气阻塞的病变均会导致通气灌注不匹配。大部分PCH患者V/Q显像基本正常,7%~10%的PCH患者可出现与慢性栓塞性肺动脉高压类似的局部区域通气灌注不匹配现象[11]。遗憾的是,我们的2例患者均未完善该项检查,考虑与诊断及鉴别诊断意识不足有关。文献回顾中完善核素V/Q显像15例。
3.2.7 右心导管检查
右心导管(right heart catheterization,RHC)检查是诊断肺动脉高压的金标准,将导管经外周静脉送入右心及肺动脉,进行血流动力学及氧动力学检测的导管技术,其中使用Swan-Ganz导管进行检查的技术称为右心漂浮导管。PCH的血流动力学符合毛细血管前肺动脉高压特征,表现为平均肺动脉压升高,肺动脉楔压正常或减低[12]。我们的2例患者都进行了右心漂浮导管检查,检查结果均符合毛细血管前肺动脉高压,文献回顾中完善RHC检查50例。RHC检查作为一种精准测量肺动脉压力的侵入性检查手段,可以通过测定血流动力学指标,明确评估肺动脉高压严重程度,提示肺动脉高压病因及分类,但考虑到其是有创操作,存在一定风险,以及检查费用等经济因素,非必要,不建议多次、重复进行RHC检查。
3.2.8 支气管镜检查
部分PCH患者在发病阶段会出现咯血症状,甚至极少数患者以咯血为首发症状。而咯血涉及诸多病因,需要充分鉴别。其中,支气管镜作为专科常规诊疗技术,在咯血诊断和鉴别诊断中的作用不容小觑[13]。支气管镜术应用指南推荐,对于不明原因咯血持续1周以上的病人,即使影像学未见明显异常,仍应行支气管镜检查以明确出血部位和原因。疑似PCH患者进行支气管镜检查,观察肺泡腔有无出血,BALF中是否存在含铁血黄素细胞对PCH诊断有一定价值。我们的2例患者均进行了支气管镜、TBLB检查,但均未能提供对最终诊断有重要价值的特异性发现,考虑与TBLB取材少、标本体积小、盲检取材部位难把控等有关。文献回顾中完善支气管镜检查11例。对PCH患者可考虑经支气管镜活检,但鉴于患者高肺动脉压力,术前需要综合评估有创检查的必要性,为降低风险建议由经验丰富的医生谨慎操作,同时做好相应预案、包括预置封堵球囊等。
3.2.9 基因检测
真核翻译起始因子2α激酶4(eukaryotic translation initiation factor 2-alpha kinase 4,EIF2AK4)基因编码GCN2(general control nonderepressible 2)激酶,可以调节细胞生长所需蛋白质的合成和代谢过程。若该基因突变可能会导致细胞不受控制地增殖。Best等[14]使用全外显子组测序方法在家族性PCH患者和散发性PCH患者均发现了EIF2AK4基因突变。国内外指南均推荐疑似PCH患者应进行基因检测,存在双等位基因突变的患者即可确诊。文献回顾中有6例患者通过结合EIF2AK4基因突变阳性、血流动力学指标和影像学表现确诊。因此,在临床诊治过程中,对疑似PCH的年轻患者推荐常规进行基因检测,尤其是临床表现不典型,没有病理检查支撑诊断的情况下,基因检测的作用至关重要。
3.2.10 组织病理学检查
2021年中国肺动脉高压诊断与治疗指南提出,PCH的诊断需要结合临床表现、基因检测、肺功能和血流动力学指标、影像学特征,必要时需要组织病理学诊断[15]。典型PCH病理特征是沿肺泡壁的毛细血管异常扩张和增生,边界清晰,毛细血管内皮细胞呈层状、结节状或片状广泛增生,可侵犯小血管和支气管,肺间质可见肺小动脉代偿性肌化,有时肺泡间隙满是具有含铁血黄素的巨噬细胞。免疫组化结果示肺泡壁内增生细胞呈CD31(+)、CD34(+)、SMA(+),其中CD31、CD34阳性表达与内皮细胞的分化密切相关,肺动脉中膜肥大梭形细胞SMA阳性[16]。我们的2例患者均存在上述典型PCH病理特征。文献回顾中完善组织病理学检查81例,患者肺泡间隙可见具有含铁血黄素的巨噬细胞25例。考虑到患者无法耐受肺活检,且PCH为罕见病,临床认识水平有限,贻误诊治致病情恶化、死亡的情况屡见不鲜,在文献回顾中超1/4的患者通过尸检获得病理诊断。
3.3 鉴别诊断
根据以往文献报道汇总,该病极易误诊为特发性肺动脉高压、肺静脉闭塞病、肺纤维化、过敏性肺炎、肺结节病、含铁血黄素沉着症或肺血栓栓塞性疾病等。本文2例患者先前分别被误诊为过敏性肺炎和弥漫性肺泡出血综合征。
3.3.1 过敏性肺炎(hypersensitivity pneumonitis,HP)
过敏性肺炎通常是易感个体吸入致敏抗原由免疫反应介导引起肺实质或气道炎症或纤维化的疾病。诊断HP可以根据详细的暴露史识别潜在致敏抗原,疾病发作时出现呼吸困难等非特异性症状,肺功能检查提示限制性通气功能障碍,影像学特征表现为磨玻璃影、正常肺组织影和空气潴留导致的马赛克衰减影混合存在,BALF示淋巴细胞计数>20%,再结合经TBLB取得的组织病理得出,极少情况需要手术肺活检。
3.3.2 弥漫性肺泡出血(Diffuse Alveolar Hemorrhage,DAH)综合征
DAH综合征是指在不同病因作用下,以肺泡毛细血管基底膜广泛破坏,终末细支气管以远的腺泡内广泛出血,充满含铁血黄素的巨噬细胞在间质内堆积为特征的临床综合征。临床诊治过程中,痰中带血、咯血、呼吸困难的临床表现,联合进行性贫血的实验室检查、弥漫性双侧肺泡浸润的影像学表现,对该病具有指向性意义。需要注意的是,DAH综合征只是一种病理现象,还需要详细地询问病史、体格检查和进一步完善其他检查,以明确诊断导致DAH综合征发生的具体病因。
3.4 治疗及预后
针对PCH的治疗措施中,肺移植是唯一能提高PCH患者生存获益的手段,例1患者确诊4个月后行肺移植手术,例2患者规律服用降肺动脉压药物,目前2例患者症状稳定,肺动脉压力明显降低。文献回顾中,22例PCH患者行肺移植手术,截至目前仅有1例患者行肺移植术后仍有双肺复发的报道。
PCH患者使用PAH靶向药物的疗效存在显著差异,部分患者使用内皮素受体拮抗剂(如波生坦)、磷酸二酯酶-5抑制剂(如西地那非)或前列环素等肺血管舒张剂后能有效改善其血流动力学,缓解临床症状,但治疗效果短暂,缺乏长期稳定性。而部分PCH患者使用上述药物可能会引起难治性肺水肿,从而发生心力衰竭导致死亡[17-18]。2022年ESC/ERS指南认为氧疗、利尿剂和PAH靶向药物的使用需因人而异。此外,有临床研究已表明干扰素-α和血管生成抑制剂伊马替尼对部分患者有一定的治疗作用,但其具体机制和适用范围仍需通过大量的临床试验探索[19-20]。需要注意的是,继发于CTD或免疫系统性疾病的PCH需要防治原发病的快速进展,具体治疗方案(如激素、免疫抑制剂等)也需结合风湿免疫专科医生的建议。国内外指南一致推荐患者一经确诊PCH后应尽早评估肺移植的可能性,同时建议PCH患者转诊至PH中心接受进一步治疗和管理。
3.5 总结和展望
PCH作为一种罕见的肺动脉高压疾病,进展迅速,诊断困难,需要综合多项临床资料,包括临床表现、基因检测、肺功能和血流动力学指标、影像学特征以及组织病理学检查等,其中组织病理诊断对确诊该疾病具有决定性意义,本文2例患者均是通过外科胸腔镜下肺组织活检术确诊的。随着人们对基因检测技术的认知不断提升,使得PCH的诊断手段更加全面且患者能凭借EIF2AK4双等位基因突变阳性直接确诊。综上所述,熟悉该病的疾病特征、提高诊断意识、早期识别并正确诊断,最终制定行之有效的治疗方案对PCH患者意义深远。
利益冲突:本研究不涉及任何利益冲突。
参考文献略。
第一作者

蔡敏
专业型硕士研究生,就读于南京大学;专业方向:内科学-呼吸与危重症医学科-间质性肺疾病。
通讯作者

张英为
主任医师,博士,副教授,硕士研究生导师,江苏省医学会呼吸病学分会呼吸内镜和介入学组成员、江苏省医师协会呼吸医师分会基层医院呼吸疾病防治学组成员、江苏省“六大人才高峰”培养对象,主持并参与国家级课题5项,省级、市级课题各1项。2013年于德国Ruhrland klinik访学3个月,2016年-2017年于美国加州大学旧金山分校访学1年。长期从事呼吸系统疾病,尤其是肺间质疾病的的临床诊治和基础研究,在国内外核心期刊发表论文40余篇,参编专著3部。
引用本文:蔡敏, 任丽君, 夏鑫烨, 刘小琴, 徐庆庆, 程乐, 徐新运, 蔡后荣, 张英为. 肺毛细血管瘤病二例报告并文献复习. 中国呼吸与危重监护杂志, 2025, 24(5): 336-343. doi: 10.7507/1671-6205.202408023
本文转载自「中国呼吸与危重监护杂志」
原链接戳:【论著】肺毛细血管瘤病二例报告并文献复习
* 文章仅供医疗卫生相关从业者阅读参考
本文完
责编:Jerry